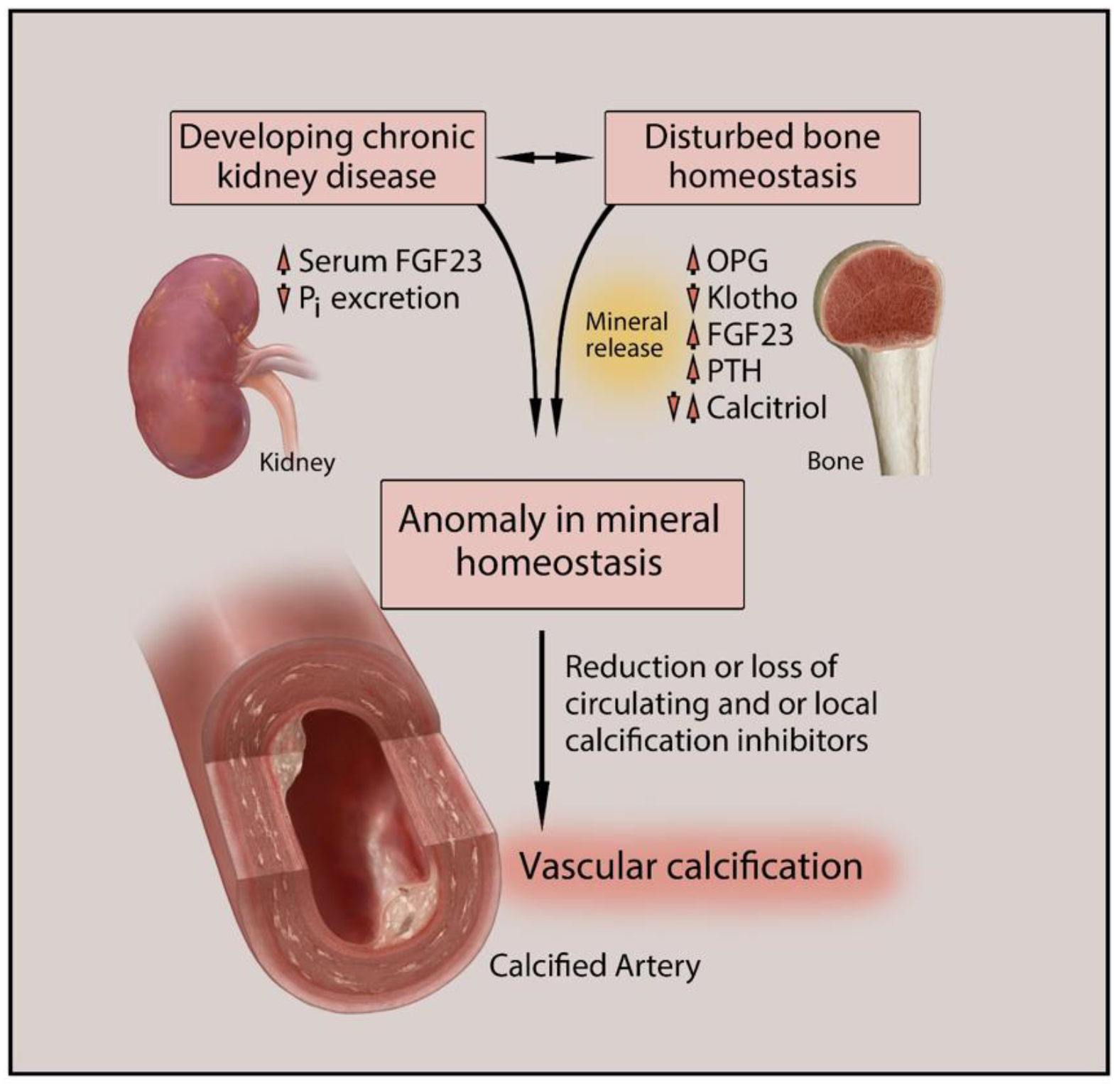
Vascular calcification (VC) is one of the major causes of cardiovascular morbidity and mortality in patients with chronic kidney disease (CKD). VC is a complex process expressing similarity to bone metabolism in onset and progression. VC in CKD is promoted by various factors not limited to hyperphosphatemia, Ca/Pi imbalance, uremic toxins, chronic inflammation, oxidative stress, and activation of multiple signaling pathways in different cell types, including vascular smooth muscle cells (VSMCs), macrophages, and endothelial cells.
- vascular calcification
- chronic kidney disease
- CKD
- uremic toxins
- hyperphosphatemia
- vascular smooth muscle cells
- VSMCs
- macrophages
- endothelium
1. Introduction
|
Promoters |
Inhibitors |
|---|---|
|
Bone morphogenetic protein (BMP) 2, 4, and 6 Osteocalcin Alkaline phosphatase (Sex determining region Y)-box 9 (SOX9) Osterix Matrix metalloproteinase (MMP) 2, 3, and 7 Runt-related transcription factor 2 (Runx2) Calcium Phosphate Glucose Advanced glycation end products Oxidized low-density lipoproteins Collagen I Receptor activator of nuclear factor-kB ligand (RANKL) |
Matrix Gla protein (MGP) Osteopontin Osteoprotegerin Vitamin K Magnesium Bone morphogenetic protein 7 (BMP7) Fetuin Klotho Parathyroid hormone (PTH) Pyrophosphate Carbonic anhydrase Collagen IV Inorganic pyrophosphate (PPi) |
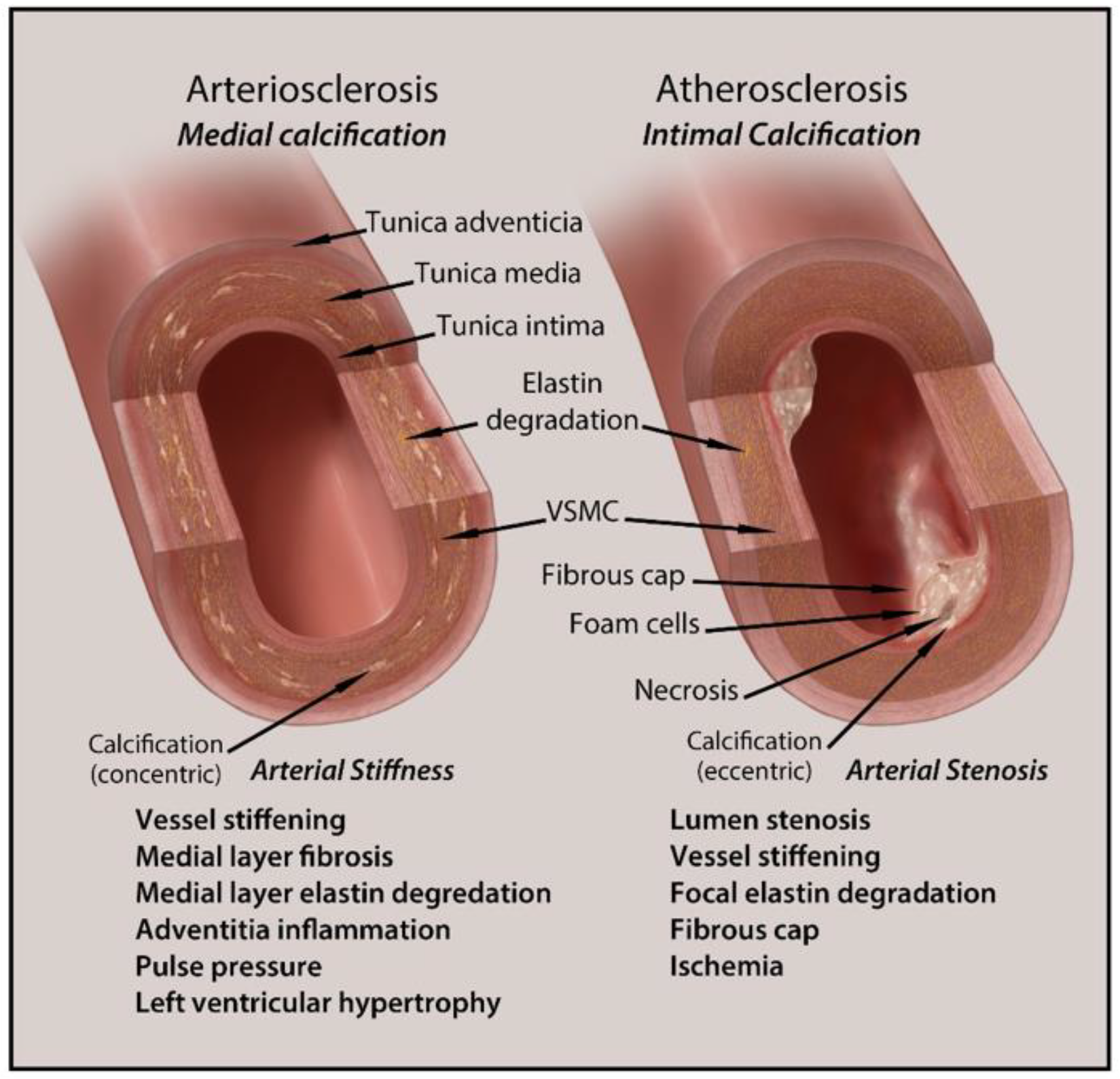
2. Types and Anatomical Presence of VC
3. Clinical Implications and Diagnosis of Vascular Calcification in CKD
4. Treatment Options for Vascular Calcification in CKD
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9040404
References
- Gao, J.; Zhang, K.; Chen, J.; Wang, M.-H.; Wang, J.; Liu, P.; Huang, H. Roles of aldosterone in vascular calcification: An update. Eur. J. Pharmacol. 2016, 786, 186–193.
- Généreux, P.; Redfors, B.; Witzenbichler, B.; Arsenault, M.-P.; Weisz, G.; Stuckey, T.D.; Rinaldi, M.J.; Neumann, F.-J.; Metzger, D.C.; Henry, T.D.; et al. Two-year outcomes after percutaneous coronary intervention of calcified lesions with drug-eluting stents. Int. J. Cardiol. 2017, 231, 61–67.
- Lee, H.Y.; Park, U.J.; Kim, H.T.; Roh, Y.-N. The Effect of Severe Femoropopliteal Arterial Calcification on the Treatment Outcome of Femoropopliteal Intervention in Patients with Ischemic Tissue Loss. Vasc. Spec. Int. 2020, 36, 96–104.
- Lacolley, P.; Regnault, V.; Laurent, S. Mechanisms of Arterial Stiffening. Arter. Thromb. Vasc. Biol. 2020, 40, 1055–1062.
- Lioufas, N.M.; Pedagogos, E.; Hawley, C.M.; Pascoe, E.M.; Elder, G.J.; Badve, S.V.; Valks, A.; Toussaint, N.D.; on behalf of the IMPROVE-CKD Investigators. Aortic Calcification and Arterial Stiffness Burden in a Chronic Kidney Disease Cohort with High Cardiovascular Risk: Baseline Characteristics of the Impact of Phosphate Reduction on Vascular End-Points in Chronic Kidney Disease Trial. Am. J. Nephrol. 2020, 51, 201–215.
- Jagieła, J.; Bartnicki, P.; Rysz, J. Selected cardiovascular risk factors in early stages of chronic kidney disease. Int. Urol. Nephrol. 2020, 52, 303–314.
- Demer, L.L.; Tintut, Y. Vascular Calcification. Circulation 2008, 117, 2938–2948.
- Chen, N.X.; Moe, S.M. Vascular calcification: Pathophysiology and risk factors. Curr. Hypertens. Rep. 2012, 14, 228–237.
- Hénaut, L.; Chillon, J.-M.; Kamel, S.; Massy, Z.A. Updates on the Mechanisms and the Care of Cardiovascular Calcification in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 233–250.
- Voelkl, J.; Lang, F.; Eckardt, K.-U.; Amann, K.; Kuro-O, M.; Pasch, A.; Pieske, B.; Alesutan, I. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell. Mol. Life Sci. 2019, 76, 2077–2091.
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular Calcification: An Update on Mechanisms and Challenges in Treatment. Calcif. Tissue Int. 2013, 93, 365–373.
- El Din, U.A.A.S.; Salem, M.M.; Azim, S.E.D.U.A. Vascular calcification: When should we interfere in chronic kidney disease patients and how? World J. Nephrol. 2016, 5, 398–417.
- Nakanishi, T.; Nanami, M.; Kuragano, T. The pathogenesis of CKD complications; Attack of dysregulated iron and phosphate metabolism. Free Radic. Biol. Med. 2020, 157, 55–62.
- Olapoju, S.O.; Adejobi, O.I.; Le Thi, X. Fibroblast growth factor 21; review on its participation in vascular calcification pathology. Vasc. Pharmacol. 2020, 125–126, 106636.
- Opdebeeck, B.; D’Haese, P.C.; Verhulst, A. Molecular and Cellular Mechanisms that Induce Arterial Calcification by Indoxyl Sulfate and P-Cresyl Sulfate. Toxins 2020, 12, 58.
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268.
- Sarnak, M.J. Cardiovascular complications in chronic kidney disease. Am. J. Kidney Dis. 2003, 41, 11–17.
- Sheng, B.; Zhu, T.; Li, J. End-stage renal disease and pulmonary hypertension. Zhong Nan Da Xue Xue Bao Yi Xue Ban = J. Cent. South Univ. Med. Sci. 2019, 44, 1419–1422.
- Arefin, S.; Buchanan, S.; Hobson, S.; Steinmetz, J.; Alsalhi, S.; Shiels, P.G.; Kublickiene, K.; Stenvinkel, P. Nrf2 in early vascular ageing: Calcification, senescence and therapy. Clin. Chim. Acta 2020, 505, 108–118.
- Lee, S.J.; Lee, I.-K.; Jeon, J.-H. Vascular Calcification—New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685.
- Thompson, B.; Towler, D.A. Arterial calcification and bone physiology: Role of the bone–vascular axis. Nat. Rev. Endocrinol. 2012, 8, 529–543.
- Xu, J.; Shi, G.-P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 2106–2119.
- Cucchiari, D.; Torregrosa, J.-V. Calcifilaxis en pacientes con enfermedad renal crónica: Una enfermedad todavía desconcertante y potencialmente mortal. Nefrología 2018, 38, 579–586.
- House, S.J.; Potier, M.; Bisaillon, J.; Singer, H.A.; Trebak, M. The non-excitable smooth muscle: Calcium signaling and phenotypic switching during vascular disease. Pflug. Arch. Eur. J. Physiol. 2008, 456, 769–785.
- Cianciolo, G.; Galassi, A.; Capelli, I.; Schillaci, R.; La Manna, G.; Cozzolino, M. Klotho-FGF23, Cardiovascular Disease, and Vascular Calcification: Black or White? Curr. Vasc. Pharmacol. 2018, 16, 143–156.
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium Regulates Key Components of Vascular Smooth Muscle Cell–Derived Matrix Vesicles to Enhance Mineralization. Circ. Res. 2011, 109, e1–e12.
- Houben, E.; Neradova, A.; Schurgers, L.J.; Vervloet, M. The influence of phosphate, calcium and magnesium on matrix Gla-protein and vascular calcification: A systematic review. G. Ital. Nefrol. 2017, 33, 1724–5590.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2009, 113, S1–S130.
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human Vascular Smooth Muscle Cells Undergo Vesicle-Mediated Calcification in Response to Changes in Extracellular Calcium and Phosphate Concentrations: A Potential Mechanism for Accelerated Vascular Calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867.
- Disthabanchong, S. Vascular calcification in chronic kidney disease: Pathogenesis and clinical implication. World J. Nephrol. 2012, 1, 43–53.
- Merjanian, R.; Budoff, M.; Adler, S.; Berman, N.; Mehrotra, R. Coronary artery, aortic wall, and valvular calcification in nondialyzed individuals with type 2 diabetes and renal disease. Kidney Int. 2003, 64, 263–271.
- Lamprea-Montealegre, J.A.; McClelland, R.L.; Astor, B.C.; Matsushita, K.; Shlipak, M.; de Boer, I.H.; Szklo, M. Chronic kidney disease, plasma lipoproteins, and coronary artery calcium incidence: The Multi-Ethnic Study of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 652.
- Kramer, H.; Toto, R.; Peshock, R.; Cooper, R.; Victor, R. Association between Chronic Kidney Disease and Coronary Artery Calcification: The Dallas Heart Study. J. Am. Soc. Nephrol. 2004, 16, 507–513.
- Qunibi, W.Y.; AbouZahr, F.; Mizani, M.R.; Nolan, C.R.; Arya, R.; Hunt, K.J. Cardiovascular calcification in Hispanic Americans (HA) with chronic kidney disease (CKD) due to type 2 diabetes. Kidney Int. 2005, 68, 271–277.
- Russo, D.; Palmiero, G.; De Blasio, A.P.; Balletta, M.M.; Andreucci, V.E. Coronary artery calcification in patients with CRF not undergoing dialysis. Am. J. Kidney Dis. 2004, 44, 1024–1030.
- Chiu, Y.-W.; Adler, S.G.; Budoff, M.J.; Takasu, J.; Ashai, J.; Mehrotra, R. Coronary artery calcification and mortality in diabetic patients with proteinuria. Kidney Int. 2010, 77, 1107–1114.
- Bellasi, A.; Raggi, P. Vascular calcification in patients with kidney disease: Techniques and Technologies to Assess Vascular Calcification. Semin. Dial. 2007, 20, 129–133.
- Uhlig, K. There Is No Practical Utility in Routinely Screening Dialysis Patients for Vascular Calcification. Semin. Dial. 2010, 23, 277–279.
- Beto, J.; Bhatt, N.; Gerbeling, T.; Patel, C.; Drayer, D. Overview of the 2017 KDIGO CKD-MBD Update: Practice Implications for Adult Hemodialysis Patients. J. Ren. Nutr. 2019, 29, 2–15.
- Ix, J.H.; Katz, R.; Kestenbaum, B.; Fried, L.F.; Kramer, H.; Stehman-Breen, C.; Shlipak, M.G. Association of Mild to Moderate Kidney Dysfunction and Coronary Calcification. J. Am. Soc. Nephrol. 2008, 19, 579–585.
- Jun, M.; Lv, J.; Perkovic, V.; Jardine, M.J. Managing cardiovascular risk in people with chronic kidney disease: A review of the evidence from randomized controlled trials. Ther. Adv. Chronic Dis. 2011, 2, 265–278.
- Himmelsbach, A.; Ciliox, C.; Goettsch, C. Cardiovascular Calcification in Chronic Kidney Disease—Therapeutic Opportunities. Toxins 2020, 12, 181.
- Jamal, S.A.; Vandermeer, B.; Raggi, P.; Mendelssohn, D.C.; Chatterley, T.; Dorgan, M.; Lok, C.E.; Fitchett, D.; Tsuyuki, R.T. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: An updated systematic review and meta-analysis. Lancet 2013, 382, 1268–1277.
- Block, G.A.; Bushinsky, D.A.; Cheng, S.; Cunningham, J.; Dehmel, B.; Drueke, T.B.; Ketteler, M.; KewalRamani, R.; Martin, K.J.; Moe, S.M.; et al. Effect of Etelcalcetide vs. Cinacalcet on Serum Parathyroid Hormone in Patients Receiving Hemodialysis with Secondary Hyperparathyroidism. JAMA 2017, 317, 156–164.
- Chitalia, N.; Recio-Mayoral, A.; Kaski, J.C.; Banerjee, D. Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis 2012, 220, 265–268.
- Perazella, M.A.; Markowitz, G.S. Bisphosphonate nephrotoxicity. Kidney Int. 2008, 74, 1385–1393.
- Bergner, R.; Diel, I.J.; Henrich, D.; Hoffmann, M.; Uppenkamp, M. Differences in Nephrotoxicity of Intravenous Bisphosphonates for the Treatment of Malignancy Related Bone Disease. Oncol. Res. Treat. 2006, 29, 534–540.
- Louvet, L.; Büchel, J.; Steppan, S.; Passlick-Deetjen, J.; Massy, Z.A. Magnesium prevents phosphate-induced calcification in human aortic vascular smooth muscle cells. Nephrol. Dial. Transplant. 2012, 28, 869–878.
- Sharples, E.J.; Pereira, D.; Summers, S.; Cunningham, J.; Rubens, M.; Goldsmith, D.; Yaqoob, M.M. Coronary artery calcification measured with electron-beam computerized tomography correlates poorly with coronary artery angiography in dialysis patients. Am. J. Kidney Dis. 2004, 43, 313–319.
- Maniscalco, B.S.; Taylor, K.A. Calcification in coronary artery disease can be reversed by EDTA–tetracycline long-term chemotherapy. Pathophysiology 2004, 11, 95–101.
- Krieger, N.S.; Frick, K.K.; Bushinsky, D.A. Mechanism of acid-induced bone resorption. Curr. Opin. Nephrol. Hypertens. 2004, 13, 423–436.
- Mendoza, F.J.; Lopez, I.; Montes de Oca, A.; Perez, J.; Rodriguez, M.; Aguilera-Tejero, E. Metabolicacidosis inhibits soft tissue calcification in uremic rats. Kidney Int. 2008, 73, 407–414.
- Simpson, C.L.; Lindley, S.; Eisenberg, C.; Basalyga, D.M.; Starcher, B.C.; Simionescu, D.T.; Vyavahare, N.R. Toward cell therapy for vascular calcification: Osteoclast-mediated demineralization of calcifiedelastin. Cardiovasc. Pathol. 2007, 16, 29–37.