Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Serena Ascrizzi | -- | 2581 | 2023-08-21 12:25:53 | | | |
| 2 | Catherine Yang | Meta information modification | 2581 | 2023-08-22 02:55:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ascrizzi, S.; Arillotta, G.M.; Grillone, K.; Caridà, G.; Signorelli, S.; Ali, A.; Romeo, C.; Tassone, P.; Tagliaferri, P. Lynch Syndrome (LS). Encyclopedia. Available online: https://encyclopedia.pub/entry/48270 (accessed on 23 July 2026).
Ascrizzi S, Arillotta GM, Grillone K, Caridà G, Signorelli S, Ali A, et al. Lynch Syndrome (LS). Encyclopedia. Available at: https://encyclopedia.pub/entry/48270. Accessed July 23, 2026.
Ascrizzi, Serena, Grazia Maria Arillotta, Katia Grillone, Giulio Caridà, Stefania Signorelli, Asad Ali, Caterina Romeo, Pierfrancesco Tassone, Pierosandro Tagliaferri. "Lynch Syndrome (LS)" Encyclopedia, https://encyclopedia.pub/entry/48270 (accessed July 23, 2026).
Ascrizzi, S., Arillotta, G.M., Grillone, K., Caridà, G., Signorelli, S., Ali, A., Romeo, C., Tassone, P., & Tagliaferri, P. (2023, August 21). Lynch Syndrome (LS). In Encyclopedia. https://encyclopedia.pub/entry/48270
Ascrizzi, Serena, et al. "Lynch Syndrome (LS)." Encyclopedia. Web. 21 August, 2023.
Copy Citation
Lynch syndrome (LS), also known as Hereditary Non-Polyposis Colorectal Cancer (HNPCC), is an autosomal dominant cancer syndrome which causes about 2–3% of cases of colorectal carcinoma. The development of LS is due to the genetic and epigenetic inactivation of genes involved in the DNA mismatch repair (MMR) system, causing an epiphenomenon known as microsatellite instability (MSI).
Lynch syndrome
hereditary non-polyposis colorectal cancer
HNPCC
1. Genetic and Molecular Background
The main molecular events underlying LS are defects in the MMR system, which is a mechanism that prevents the accumulation of mutations due to DNA mismatch that may occur during cell division, including single nucleotide mismatches, insertions, and deletion loops [1][2]. The MMR system functions as a multienzyme complex capable of forming heterodimers to repair DNA damage and promote genomic stability [3][4] with high fidelity and efficiency [5].
MMR multi-enzyme complexes mainly include MSH2 and MLH1 proteins. These two proteins form protein heterodimers with other players. MSH2 associates with MSH6 or MSH3 (MutS α or MutS β); MLH1 associates with PMS2, PMS1, MLH3 (MutL α, MutL β, MutL γ) [6].
The process of damage repair can include three steps. The first step begins with the identification of DNA mismatch by the MutS and MutL complexes. Once the complexes are formed and the damaged region is recognized, the initiation complex is assembled, which consists of MutS α or β, MutL α (MLH1-PMS2), and PCNA (Figure 1A) [7]. EXO-1, DNA polymerase δ (Figure 1B), and DNA ligase are recruited to this complex, which make the cut in the damage sequence, correctly resynthesize the strand, and reconstitute the base bonds, respectively (Figure 1C) [8].
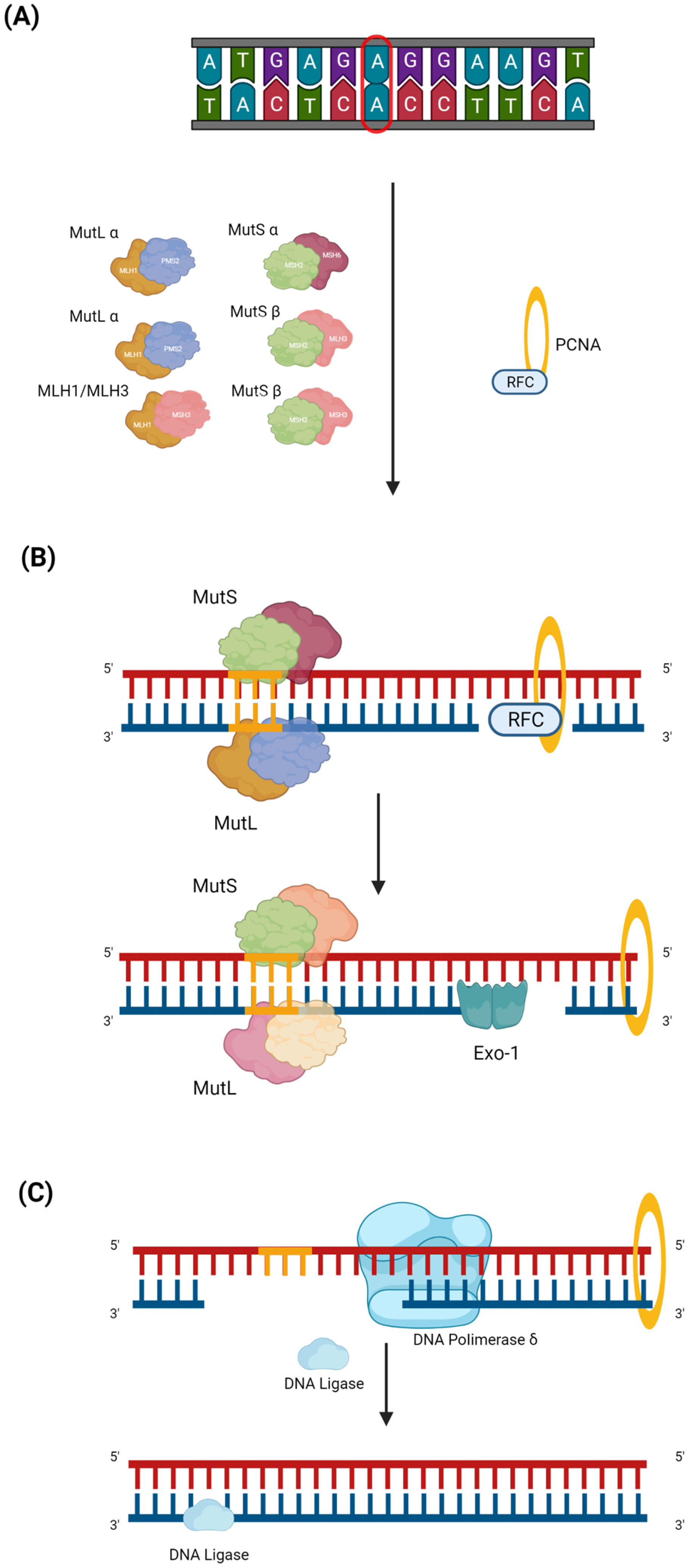
Figure 1. MMR pathway. Mismatch repair (MMR) is a DNA repair mechanism that is activated during the replication phase. Despite the high replication fidelity of polymerases, base insertion errors can be made, creating single nucleotide mismatches, insertions, and deletion loops in repeated stretches of DNA, e.g., in the microsatellite region. The MMR can be divided into three steps. (A) The first is the damage recognition step by the MutS complex, which consists of MutS α (MSH2 and MSH6) or MutS β (MSH2 and MSH3). After recognizing the damaged region, the MutL (MutL α (MLH1 and PMS2), MutL β (MLH1 and PMS1), or MutL γ (MLH1 and MLH3)) complex is formed. The MutS and MutL heterodimeric complexes bind to the damaged strand region, and recruiting PCNA is transported by RFC, which, once it has bound the DNA, is ejected. (B) During the second step, EXO-1, an enzyme belonging to exonucleases which acts to mediate base excision, is recruited. (C) In the last step, first, DNA polymerase δ, which latches onto the PCNA closed around the DNA, intervenes to properly synthesize the mismatch; then, DNA ligase 1 resynthesizes the bonds between the bases.
As previously observed, when the MMR system is defective, there is an accumulation of mutations, which causes the microsatellite instability epiphenomenon (MSI-H) and prompts the onset of several cancers, including LS [3][6].
Mutations in MLH1 and MSH2 are estimated to cause about 70% of LS cases [9]; MSH6 mutations cause up to 14%, and PMS2 mutations contribute to less than 15% [10]. The germinal deletion of EPCAM, a gene not directly belonging to MMR, inactivates MSH2, causing LS in 1–3% of cases [11][12]. However, the number of reported mutations affecting MMR genes varies considerably [13] because LS cases are diagnosed in the background of cancers whose variants may be different in terms of number, type, and the genes involved [14]. Mutations in MLH1 and MSH2 [15] are associated with a high risk of developing LS; mutations in MSH6 are associated with intermediate risk, and mutations in PMS2 are associated with low risk [16]. In the case of EPCAM, the presence of mutations are associated with a high risk of CRC development but low risk of extra-colic cancer development [17][18]. In any case, the overall risk of developing cancer in LS patients also depends on gender, age, history of malignancy, environmental factors, and lifestyle [13].
The malignant transformation in LS-related CRC has been described as a multistep process and explained by different models of progression (Figure 2). The first proposed model supposes that tumors arise from polypoid lesions with proficient MMR, whose deficiency will occur at later steps [19]. In particular, it was assumed that the formation of sporadic polyps with dysfunctional APC-mediated systems was the event promoting the adenoma to CRC formation and that the loss of MMR, promoting the accumulation of somatic mutations, was a subsequent event underlying the genesis of the invasive form (Figure 2A) [20][21]. Advances in molecular analysis technologies have led to the definition of a new model that completely overcomes adenomatous precursors to explain LS-associated CRC [22]. This new model is based on the detection of intestinal crypt foci with dMMR [23]. These foci are histologically normal and apparently non-neoplastic, which is common in the intestinal epithelium of healthy LS carriers without cancer. These crypts are adjacent to others with a functional dMMR, suggesting a role of this system in the adenoma initiation. These areas may acquire somatic mutations in TP53 or CTNNB1 by causing immediate and direct invasive tumor growth (Figure 2B) [24]. This model is able to explain the accelerated development of CRCs within a short interval between screening colonoscopies [25].
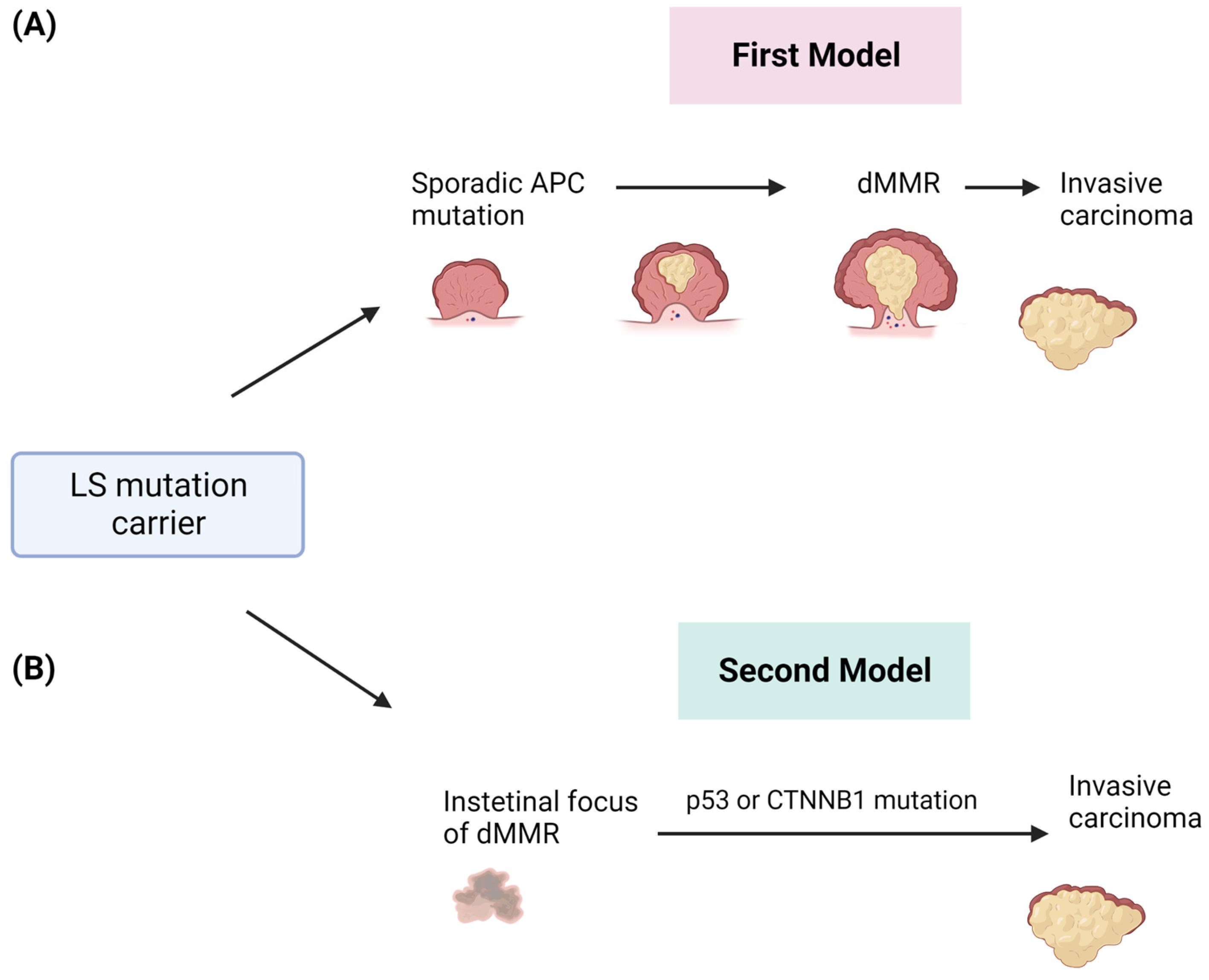
Figure 2. Two different models of CRC development in patients affected by LS. (A) The first model involves the sporadic acquisition of early APC gene mutation in adenomas and after the onset of dMMR and invasive tumors. (B) The second model describes the early onset of dMMR and the direct development of invasive tumors.
2. Clinical and Diagnostic Overview
From a pathological point of view, LS-related CRC is characterized by low differentiation (grade 4), mucinous or castellated ring cells, and a bone marrow growth pattern with rich Crohn-type lymphocyte infiltrates, representing a response to neoantigens generated by the high mutational burden [19][26][27].
From a genetic perspective [28], the phenotypic histopathological and clinical manifestations of LS mainly depend on the mutated gene responsible for MMR. For example, individuals with MSH2 mutations frequently develop genito–urinary tract and endometrial neoplasms [29], while individuals with MSH6 mutation are more prone to develop Breast Cancer (BC) [30]. Therefore, surveillance and treatment programs for these patients should be modulated based on the mutated genes [31].
CRC in patients with LS has different features than sporadic CRC [32]. Usually LS-related CRC localizes to the right/proximal side of the colon, manifest early (40–50 years old), and have propensity for synchronous and metachronous colorectal carcinoma, again according to genotypes [33]. For example, LS patients with PSM2 mutations [34] develop malignancies later than other patients, while patients with MSH6 mutations have CRC and endometrial carcinoma later in life compared with MLHI or MSH2 mutation carriers [15].
To date, early diagnosis in individuals with typical LS genetic mutation has a triple significance as (i) it allows for the development of a program of close surveillance of the patient themselves in view of their high risk of developing cancer; (ii) it is necessary to offer their family members appropriate screening checks; and, above all, (iii) it has proved to be essential because of the known therapeutic implications (avoid chemotherapy treatment with adjuvant purposes and the use of immune checkpoint inhibitors) [35][36].
In the past, both clinical and histopathological criteria have been used to diagnose LS [37]. The original clinical criteria were the Amsterdam I criteria, according to which the clinical diagnosis of LS could be made if the following conditions were met: the presence of three family members diagnosed with CRC in two successive generations (with one first-degree relative from the other two), at least one case of a family member with CRC before the age of 50, and the exclusion of familial adenomatous polyposis [38]. These criteria were modified in 1999 to include neoplasms of the extra-colonic district in the Amsterdam II criteria [39].
In addition to these purely clinical criteria, the Bethesda criteria were devised [40], which combine family history with histopathologic features typical of LS-related malignancies (mucinous histology, castellated ring cells, presence of abundant lymphocytic infiltrates, Crohn-like reactions, bone marrow growth pattern): CRC diagnosis at under 50 years, the presence of synchronous or metachronous colon cancers, diagnosis of colon cancer at 60 years of age but histologic features similar to MSI-H, at least one first-degree relative with Lynch-related cancer and/or CRC diagnosed when the relative is under 50 years of age, and two or more first- or second-degree relatives with Lynch-related cancers [41].
New risk prediction algorithms [37] such as PREMM [42][43][44], MMRpro [44], and MMRpredict [45] have recently been devised for LS. These quantify, in percentage, the risk of a patient carrying a mutation in the LS genes, therefore determining the need for genetic evaluation and possible germline genetic testing based on the patient’s personal and family history of cancer [31].
Notably, PREMM5 is the first clinical model based on the patient’s sex, age, and personal/family history to provide a risk assessment for all five LS genes (MSH2, EPCAM, MLH1, MSH6, and PMS2); a score of 2.5% or higher indicates a need for genetic evaluation [46].
However, all these clinical patterns of diagnosis have a very low detection rate. Less than 50% of LS patients meet these criteria [47]. This is due to limited knowledge and difficulty in reconstructing family history [26] but also due to wanting to identify higher-risk individuals with these criteria rather than the entire spectrum of LS patients [48].
Bethesda criteria combining clinical features with histopathologic features also demonstrated high sensitivity but low specificity [37].
Today, the diagnosis of LS is performed through an early evaluation of tumor tissue or by immunohistochemical analysis to demonstrate dMMR but also by using a polymerase chain reaction (PCR)-based test to demonstrate MSI status [26].
The results of these tests have been shown to be highly concordant [26], allowing for the identification of patients to be tested to investigate the germline variants of the selected genes involved in MMR [35].
The PCR test for MSI evaluation compares changes in short repeated sequences at different loci (usually at the five most frequent loci) [36]. The tumor tissue sample is considered MSI-High (MSI-H) if MSI is detected in at least two or more out of five loci (30%), MSI-Low (MSI-L) if MSI is detected in less than one out of five loci (less than 30%), and microsatellite stability (MSS) is deemed to have occurred if MSI is not detected in any of the loci [36][49]. MSI-H indicates a dMMR, while MSI-L or no MSI indicates efficient MMR repair [26]. Clinically, MSI-L and MSS behave similarly and are often clustered together [37].
However, it should be noted that most cancers with MSI-H are attributable to the somatic and/or epigenetic inactivation of genes involved in MMR rather than germline mutations [36]. Only 15% of CRC cases with MSI-H are due to LS [50]. The most frequent somatic mutation of MMR genes that results in MSI-H with CRC and endometrial development is promoter hypermethylation, which results in MLH1 silencing [36][51]. Since MLH1 and PMS2 proteins function as a stable heterodimer, the simultaneous loss of expression of both means that there is an underlying alteration of MLH1 caused by the somatic methylation of the MLH1 promoter (sporadic case) or by the germline mutation of MLH1 (LS) [26].
Considering the high-frequency somatic hypermethylation of MLH1 in CRC and endometrial cases, before germline testing and genetic evaluation, hypermethylation of the MLH1 promoter should be excluded either by direct evaluation or by assessing the presence of the somatic BRAF V600E mutation, which, as a result, acquires negative predictive values in the case of CRC [36][52].
However, compared with MLH1, the loss of expression of MSH2 or MSH6 (or both) and PMS2 is usually caused by a germline mutation. Therefore, patients with CRC and a loss of MLH1 with the absence of the BRAF V600E mutation or MLH1 hypermethylation, as well as patients with a loss of MSH2 or MSH6 (or both) or PMS2 and elevated MSI, should be referred for genetic counseling and germline genetic testing to confirm LS [26].
In the past few years, patients with colorectal cancer or MSI-H/dMMR endometrial carcinoma without germline mutations and MLH1 promoter hypermethylation were erroneously referred to as “presumed LS”. Nowadays, due to the development and increasing use of next-generation sequencing (NGS) on tumor tissue in clinical–diagnostic practice to determine MMR status [53], they are instead referred to as “Lynch-like syndrome” [26]. Evidence of dMMR/MSI-H due to the biallelic somatic mutations of MMR genes or single somatic mutations with heterozygous loss of the other allele have been reported [54].
The identification of Lynch-like syndrome has a very important therapeutic implication since these patients can benefit from treatment with immune checkpoint inhibitors [26].
NGS detects tumor mutational burden (TMB), also identifying therapeutic targets, as in the case of colorectal mutations affecting genes involved in the RAS pathway [26]. When NGS detects a suspected germline mutation, confirmatory sequencing of the germline should be performed via orthogonal sequencing [26]. Today, NGS is increasingly used because its multigene panels offer numerous advantages, such as the following: identifying individuals with LS and atypical clinical phenotypes, identifying mutations in genes associated with the risk of developing cancer, identifying target mutations but also having lower cost content, and speed in the availability of results [55].
Recent data also show that somatic NGS panels have concordant results with traditional immunohistochemical and molecular PCR assays for the evaluation of MSI/dMMR [56]. These NGS panels also seem to overcome some of the biases of the PCR assay, and it has been suggested that they may replace them in the future, especially when implemented for the detection of other relevant somatic mutations [55].
Although it is not yet widespread clinical practice, regardless of the test used, a screening for LS should be performed whenever a new cancer diagnosis is made for therapeutic implications, for the prevention of further cancer in the same patient, and for the prevention of primary cancers in the patient’s family members [36].
Today, guidelines recommend (i) screening all diagnosed colorectal and endometrial cancer patients for LS; (ii) excluding hypermethylation of the MLH1 promoter, MMR-D/MSI-H, CRC, and/or BRAF V6OOE mutation in the case of MLH1 and PMS2 expression; and (iii) offering patients genetic evaluations and possible confirmatory germline testing.
3. LS and Breast Cancer: What Is the Real Link?
Breast cancer (BC) is one of the cancers associated with LS, as reported by some studies suggesting that women with LS may have a two- to three-fold increased risk of developing BC than the general population.
To date, the relationship between LS and BC remains much debated. The available data lack statistical significance because of the small sample sizes, the type of retrospective studies, and the fact that only epidemiological analyses are available [57]. However, the onset of BC in LS patients is around 50 years of age; therefore, mammography is still strongly recommended as a screening technique for the general population [58].
In patients with LS with BC, genetic testing and counseling are critical for estimating the risk of developing other cancers and choosing appropriate screening and management strategies. It would be interesting to more thoroughly investigate the link between BC and LS in order to correlate them with a unique BC histotype and/or phenotype (Her2+? Triple negative?). It should be mentioned that a recent molecular approach has demonstrated an increased risk of developing BC in patients with LS secondary to MSH6 and PMS2 germline mutations [30].
Moreover, the potential relationship between LS-BC and MSI could confirm an already established therapeutic strategy [59]. As a matter of fact, MSI is recognized as a predictive biomarker of response to immunotherapy [60]. Therefore, immunohistochemical testing for MSI on tumor tissue could also be considered in the case of BC.
While regular screening and genetic counseling can help manage risk and prevention strategies for people with LS and BC, the characterization of these patients’ MMR status is difficult due to the absence of tumor-specific guidelines and/or companion diagnostic tests. Therefore, accurately identifying dMMR-BC can be a challenge [61].
References
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710.
- Rustgi, A.K. The genetics of hereditary colon cancer. Genes. Dev. 2007, 21, 2525–2538.
- De’Angelis, G.L.; Bottarelli, L.; Azzoni, C.; De’Angelis, N.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite instability in colorectal cancer. Acta Biomed. 2018, 89, 97–101.
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. Rev. Mutat. Res. 2017, 773, 174–187.
- Edelbrock, M.A.; Kaliyaperumal, S.; Williams, K.J. DNA mismatch repair efficiency and fidelity are elevated during DNA synthesis in human cells. Mutat. Res. 2009, 662, 59–66.
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162.
- Guo, J.; Chen, L.; Li, G.M. DNA mismatch repair in trinucleotide repeat instability. Sci. China Life Sci. 2017, 60, 1087–1092.
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374.
- Dowty, J.G.; Win, A.K.; Buchanan, D.D.; Lindor, N.M.; Macrae, F.A.; Clendenning, M.; Antill, Y.C.; Thibodeau, S.N.; Casey, G.; Gallinger, S.; et al. Cancer risks for MLH1 and MSH2 mutation carriers. Hum. Mutat. 2013, 34, 490–497.
- Palomaki, G.E.; McClain, M.R.; Melillo, S.; Hampel, H.L.; Thibodeau, S.N. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet. Med. 2009, 11, 42–65.
- Kempers, M.J.; Kuiper, R.P.; Ockeloen, C.W.; Chappuis, P.O.; Hutter, P.; Rahner, N.; Schackert, H.K.; Steinke, V.; Holinski-Feder, E.; Morak, M.; et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: A cohort study. Lancet Oncol. 2011, 12, 49–55.
- Weissman, S.M.; Burt, R.; Church, J.; Erdman, S.; Hampel, H.; Holter, S.; Jasperson, K.; Kalady, M.F.; Haidle, J.L.; Lynch, H.T.; et al. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline. J. Genet. Couns. 2012, 21, 484–493.
- Cerretelli, G.; Ager, A.; Arends, M.J.; Frayling, I.M. Molecular pathology of Lynch syndrome. J. Pathol. 2020, 250, 518–531.
- Seth, S.; Ager, A.; Arends, M.J.; Frayling, I.M. Lynch syndrome—Cancer pathways, heterogeneity and immune escape. J. Pathol. 2018, 246, 129–133.
- Bonadona, V.; Bonaiti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310.
- Senter, L.; Clendenning, M.; Sotamaa, K.; Hampel, H.; Green, J.; Potter, J.D.; Lindblom, A.; Lagerstedt, K.; Thibodeau, S.N.; Lindor, N.M.; et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 2008, 135, 419–428.
- Lynch, H.T.; Riegert-Johnson, D.L.; Snyder, C.; Lynch, J.F.; Hagenkord, J.; Boland, C.R.; Rhees, J.; Thibodeau, S.N.; Boardman, L.A.; Davies, J.; et al. Lynch syndrome-associated extracolonic tumors are rare in two extended families with the same EPCAM deletion. Am. J. Gastroenterol. 2011, 106, 1829–1836.
- Grandval, P.; Baert-Desurmont, S.; Bonnet, F.; Bronner, M.; Buisine, M.P.; Colas, C.; Noguchi, T.; North, M.O.; Rey, J.M.; Tinat, J.; et al. Colon-specific phenotype in Lynch syndrome associated with EPCAM deletion. Clin. Genet. 2012, 82, 97–99.
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Oliveira, D.M.; Laudanna, C.; Migliozzi, S.; Zoppoli, P.; Santamaria, G.; Grillone, K.; Elia, L.; Mignogna, C.; Biamonte, F.; Sacco, R.; et al. Identification of different mutational profiles in cancers arising in specific colon segments by next generation sequencing. Oncotarget 2018, 9, 23960–23974.
- Yurgelun, M.B.; Goel, A.; Hornick, J.L.; Sen, A.; Turgeon, D.K.; Ruffin, M.T.t.; Marcon, N.E.; Baron, J.A.; Bresalier, R.S.; Syngal, S.; et al. Microsatellite instability and DNA mismatch repair protein deficiency in Lynch syndrome colorectal polyps. Cancer Prev. Res. 2012, 5, 574–582.
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Blaker, H.; et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int. J. Cancer 2018, 143, 139–150.
- Kloor, M.; Huth, C.; Voigt, A.Y.; Benner, A.; Schirmacher, P.; von Knebel Doeberitz, M.; Blaker, H. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: A pathological study. Lancet Oncol. 2012, 13, 598–606.
- Ahadova, A.; von Knebel Doeberitz, M.; Blaker, H.; Kloor, M. CTNNB1-mutant colorectal carcinomas with immediate invasive growth: A model of interval cancers in Lynch syndrome. Fam. Cancer 2016, 15, 579–586.
- Moller, P.; Seppala, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: A report from the prospective Lynch syndrome database. Gut 2017, 66, 1657–1664.
- Sinicrope, F.A. Lynch Syndrome-Associated Colorectal Cancer. N. Engl. J. Med. 2018, 379, 764–773.
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology 2008, 134, 988–997.
- Barrow, E.; Hill, J.; Evans, D.G. Cancer risk in Lynch Syndrome. Fam. Cancer 2013, 12, 229–240.
- Win, A.K.; Lindor, N.M.; Young, J.P.; Macrae, F.A.; Young, G.P.; Williamson, E.; Parry, S.; Goldblatt, J.; Lipton, L.; Winship, I.; et al. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J. Natl. Cancer Inst. 2012, 104, 1363–1372.
- Roberts, M.E.; Jackson, S.A.; Susswein, L.R.; Zeinomar, N.; Ma, X.; Marshall, M.L.; Stettner, A.R.; Milewski, B.; Xu, Z.; Solomon, B.D.; et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet. Med. 2018, 20, 1167–1174.
- Biller, L.H.; Syngal, S.; Yurgelun, M.B. Recent advances in Lynch syndrome. Fam. Cancer 2019, 18, 211–219.
- Vasen, H.F. Clinical description of the Lynch syndrome . Fam. Cancer 2005, 4, 219–225.
- Kalady, M.F.; McGannon, E.; Vogel, J.D.; Manilich, E.; Fazio, V.W.; Church, J.M. Risk of colorectal adenoma and carcinoma after colectomy for colorectal cancer in patients meeting Amsterdam criteria. Ann. Surg. 2010, 252, 507–511; discussion 11-3.
- Ten Broeke, S.W.; van der Klift, H.M.; Tops, C.M.J.; Aretz, S.; Bernstein, I.; Buchanan, D.D.; de la Chapelle, A.; Capella, G.; Clendenning, M.; Engel, C.; et al. Cancer Risks for PMS2-Associated Lynch Syndrome. J. Clin. Oncol. 2018, 36, 2961–2968.
- Kanth, P.; Grimmett, J.; Champine, M.; Burt, R.; Samadder, N.J. Hereditary Colorectal Polyposis and Cancer Syndromes: A Primer on Diagnosis and Management. Am. J. Gastroenterol. 2017, 112, 1509–1525.
- Yurgelun, M.B.; Hampel, H. Recent Advances in Lynch Syndrome: Diagnosis, Treatment, and Cancer Prevention. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 101–109.
- Cohen, S.A.; Pritchard, C.C.; Jarvik, G.P. Lynch Syndrome: From Screening to Diagnosis to Treatment in the Era of Modern Molecular Oncology. Annu. Rev. Genom. Hum. Genet. 2019, 20, 293–307.
- Vasen, H.F.; Mecklin, J.P.; Khan, P.M.; Lynch, H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis. Colon Rectum 1991, 34, 424–425.
- Vasen, H.F.; Watson, P.; Mecklin, J.P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999, 116, 1453–1456.
- Rodriguez-Bigas, M.A.; Boland, C.R.; Hamilton, S.R.; Henson, D.E.; Jass, J.R.; Khan, P.M.; Lynch, H.; Perucho, M.; Smyrk, T.; Sobin, L.; et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: Meeting highlights and Bethesda guidelines. J. Natl. Cancer Inst. 1997, 89, 1758–1762.
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Ruschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268.
- Kastrinos, F.; Steyerberg, E.W.; Balmana, J.; Mercado, R.; Gallinger, S.; Haile, R.; Casey, G.; Hopper, J.L.; LeMarchand, L.; Lindor, N.M.; et al. Comparison of the clinical prediction model PREMM(1,2,6) and molecular testing for the systematic identification of Lynch syndrome in colorectal cancer. Gut 2013, 62, 272–279.
- Kastrinos, F.; Steyerberg, E.W.; Mercado, R.; Balmana, J.; Holter, S.; Gallinger, S.; Siegmund, K.D.; Church, J.M.; Jenkins, M.A.; Lindor, N.M.; et al. The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology 2011, 140, 73–81.
- Chen, S.; Wang, W.; Lee, S.; Nafa, K.; Lee, J.; Romans, K.; Watson, P.; Gruber, S.B.; Euhus, D.; Kinzler, K.W.; et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA 2006, 296, 1479–1487.
- Green, R.C.; Parfrey, P.S.; Woods, M.O.; Younghusband, H.B. Prediction of Lynch syndrome in consecutive patients with colorectal cancer. J. Natl. Cancer Inst. 2009, 101, 331–340.
- Kastrinos, F.; Uno, H.; Ukaegbu, C.; Alvero, C.; McFarland, A.; Yurgelun, M.B.; Kulke, M.H.; Schrag, D.; Meyerhardt, J.A.; Fuchs, C.S.; et al. Development and Validation of the PREMM(5) Model for Comprehensive Risk Assessment of Lynch Syndrome. J. Clin. Oncol. 2017, 35, 2165–2172.
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932.
- Kastrinos, F.; Ojha, R.P.; Leenen, C.; Alvero, C.; Mercado, R.C.; Balmana, J.; Valenzuela, I.; Balaguer, F.; Green, R.; Lindor, N.M.; et al. Comparison of Prediction Models for Lynch Syndrome Among Individuals With Colorectal Cancer. J. Natl. Cancer Inst. 2016, 108, djv308.
- Hendriks, Y.M.; de Jong, A.E.; Morreau, H.; Tops, C.M.; Vasen, H.F.; Wijnen, J.T.; Breuning, M.H.; Brocker-Vriends, A.H. Diagnostic approach and management of Lynch syndrome (hereditary nonpolyposis colorectal carcinoma): A guide for clinicians. CA Cancer J. Clin. 2006, 56, 213–225.
- Guastadisegni, C.; Colafranceschi, M.; Ottini, L.; Dogliotti, E. Microsatellite instability as a marker of prognosis and response to therapy: A meta-analysis of colorectal cancer survival data. Eur. J. Cancer 2010, 46, 2788–2798.
- Tsibel, B.N.; Bochkareva, A.K. Functional morphology of adenohypophysis, thymus, and adrenal cortex in sudden infant death syndrome. Arkh. Patol. 1998, 60, 23–27.
- Adar, T.; Rodgers, L.H.; Shannon, K.M.; Yoshida, M.; Ma, T.; Mattia, A.; Lauwers, G.Y.; Iafrate, A.J.; Chung, D.C. A tailored approach to BRAF and MLH1 methylation testing in a universal screening program for Lynch syndrome. Mod. Pathol. 2017, 30, 440–447.
- Hampel, H.; Pearlman, R.; Beightol, M.; Zhao, W.; Jones, D.; Frankel, W.L.; Goodfellow, P.J.; Yilmaz, A.; Miller, K.; Bacher, J.; et al. Assessment of Tumor Sequencing as a Replacement for Lynch Syndrome Screening and Current Molecular Tests for Patients With Colorectal Cancer. JAMA Oncol. 2018, 4, 806–813.
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; de la Chapelle, A.; Pritchard, C.C. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014, 147, 1308–1316.e1.
- Boland, P.M.; Yurgelun, M.B.; Boland, C.R. Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA Cancer J. Clin. 2018, 68, 217–231.
- Nowak, J.A.; Yurgelun, M.B.; Bruce, J.L.; Rojas-Rudilla, V.; Hall, D.L.; Shivdasani, P.; Garcia, E.P.; Agoston, A.T.; Srivastava, A.; Ogino, S.; et al. Detection of Mismatch Repair Deficiency and Microsatellite Instability in Colorectal Adenocarcinoma by Targeted Next-Generation Sequencing. J. Mol. Diagn. 2017, 19, 84–91.
- Ford, J.M. Is breast cancer a part of Lynch syndrome? Breast Cancer Res. 2012, 14, 110.
- Buerki, N.; Gautier, L.; Kovac, M.; Marra, G.; Buser, M.; Mueller, H.; Heinimann, K. Evidence for breast cancer as an integral part of Lynch syndrome. Genes. Chromosomes Cancer 2012, 51, 83–91.
- Schwartz, C.J.; da Silva, E.M.; Marra, A.; Gazzo, A.M.; Selenica, P.; Rai, V.K.; Mandelker, D.; Pareja, F.; Misyura, M.; D’Alfonso, T.M.; et al. Morphologic and Genomic Characteristics of Breast Cancers Occurring in Individuals with Lynch Syndrome. Clin. Cancer Res. 2022, 28, 404–413.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Sajjadi, E.; Venetis, K.; Piciotti, R.; Invernizzi, M.; Guerini-Rocco, E.; Haricharan, S.; Fusco, N. Mismatch repair-deficient hormone receptor-positive breast cancers: Biology and pathological characterization. Cancer Cell Int. 2021, 21, 266.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Entry Collection:
Gastrointestinal Disease
Revisions:
2 times
(View History)
Update Date:
22 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No