Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Man, A.W.C.; Zhou, Y.; Xia, N.; Li, H. Perivascular Adipose Tissue Oxidative Stress in Obesity. Encyclopedia. Available online: https://encyclopedia.pub/entry/48129 (accessed on 23 July 2026).
Man AWC, Zhou Y, Xia N, Li H. Perivascular Adipose Tissue Oxidative Stress in Obesity. Encyclopedia. Available at: https://encyclopedia.pub/entry/48129. Accessed July 23, 2026.
Man, Andy W. C., Yawen Zhou, Ning Xia, Huige Li. "Perivascular Adipose Tissue Oxidative Stress in Obesity" Encyclopedia, https://encyclopedia.pub/entry/48129 (accessed July 23, 2026).
Man, A.W.C., Zhou, Y., Xia, N., & Li, H. (2023, August 16). Perivascular Adipose Tissue Oxidative Stress in Obesity. In Encyclopedia. https://encyclopedia.pub/entry/48129
Man, Andy W. C., et al. "Perivascular Adipose Tissue Oxidative Stress in Obesity." Encyclopedia. Web. 16 August, 2023.
Copy Citation
Perivascular adipose tissue (PVAT) adheres to most systemic blood vessels in the body. Healthy PVAT exerts anticontractile effects on blood vessels and further protects against cardiovascular and metabolic diseases. Healthy PVAT regulates vascular homeostasis via secreting an array of adipokine, hormones, and growth factors. Normally, homeostatic reactive oxygen species (ROS) in PVAT act as secondary messengers in various signalling pathways and contribute to vascular tone regulation.
antioxidant
reactive oxygen species
eNOS
1. Introduction
Obesity is now known as an epidemic worldwide, which has become a global public health concern and burden [1]. Obesity is a well-known risk factor for cardiovascular disorders like endothelial dysfunction, atherosclerosis, hypertension, and coronary artery disease [2]. In 1991, the pioneering work of Soltis and Cassis suggested that perivascular adipose tissue (PVAT), a functional specialised ectopic fat depot, acts as a critical modulator of vascular physiology and pathology [3]. Indeed, accumulating data from both clinical and experimental studies demonstrate that the dysfunction of PVAT is a causal link between metabolic diseases and cardiovascular complications [4][5][6]. Most blood vessels, including large arteries and veins, small and resistance vessels, and skeletal muscle microvessels, are surrounded by PVAT [7]. PVAT stays in close proximity to the tunica adventitia of blood vessels, serving as a pivotal endocrine and/or paracrine tissue that maintains cardiovascular and metabolic homeostasis. PVAT contains both white and brown adipocytes [8]. Apart from adipocytes, endothelial cells, fibroblasts, immune cells extracellular matrix, and adrenergic nerves endings are also present in PVAT. Depending on the vessel type and region, PVAT may have different compositional, phenotypic, and functional aspects throughout the vascular system [9][10]. The phenotype of PVAT has been extensively reviewed [11][12][13][14][15]. In recent decades, revealing the crosstalk between blood vessels and PVAT has become a particular interest in the field of vascular biology. Apart from its structural and mechanical roles in vascular support, PVAT is actively involved in vascular homeostasis and contributes to vascular dysfunction associated with cardiovascular and metabolic diseases.
A healthy PVAT Is known to exert anticontractile effects on blood vessels in both animal models and humans [16][17]. PVAT, as a endocrine and/or paracrine tissue, regulates vascular function by releasing various vaso-active factors, including adipokines, chemokines, cytokines, hydrogen sulphide (H2S), nitric oxide (NO), and reactive oxygen species (ROS) [7]. These vasoactive substances could enter the endothelial layer of the vessel wall by diffusion or via the vasa vasorum or the small media conduit networks that connect the media with the adventitia layer [9][18][19]. These factors produced from PVAT include proinflammatory and anti-inflammatory molecules, which take part in various cellular processes, including smooth muscle proliferation and migration, vascular tone, inflammation, and oxidative stress in the vasculature [10][20].
Depending on the ‘health status’ of PVAT, it may elicit beneficial or harmful effects on the vasculature [21]. In obesity, PVAT becomes dysfunctional and exerts detrimental effects on the blood vessels [4][5][6]. The ‘obesity triad’ is proposed as the central mechanism in obesity-induced PVAT dysfunction [7]. The obesity triad consists of the interactions among PVAT hypoxia, inflammation, and oxidative stress. Among the triad, oxidative stress is a pivotal pathophysiological process in cardiovascular and metabolic complications, including obesity, type 2 diabetes, and hypertension. As oxidative stress is a key feature of hypertension, it is also known to regulate redox-dependent inflammatory molecules [22]. Normally, homeostatic ROS act as crucial secondary messengers in different signalling pathways of both innate and adaptive immune responses [23].
2. PVAT Oxidative Stress in Obesity
Obesity is a condition of excessive fat mass and subclinical inflammation. The prevalence of obesity has doubled worldwide over the past few decades, as well as the concomitant increase in obesity-associated cardiovascular diseases [24]. Obesity is a major risk factor for cardiovascular and metabolic diseases, including type 2 diabetes, insulin resistance, and hypertension [25]. In fact, endothelial dysfunction is not always evident in obese patients in vitro, although they have a higher risk of developing hypertension, cardiomyopathy, and stroke. Various studies have demonstrated that the anti-contractile effects of PVAT are attenuated in obesity [4][26][27]. Indeed, PVAT dysfunction, but not obesity itself, plays an important role in obesity-induced vascular disorders. In mice aortas, the responses to vasodilators were not different between the aortas isolated from obese and lean mice, while vasodilator responses were attenuated in the aortas isolated from obese mice when PVAT was attached [26][28]. In addition, mesenteric arteries incubated with thoracic PVAT from HFD-fed rats showed diminished endothelium-dependent relaxation compared to those incubated with thoracic PVAT from NCD-fed rats [29]. This suggests that the detrimental effects of obesity do not directly influence the intrinsic vascular reactivity but rather the function of PVAT and PVAT dysfunction are closely related to the development of obesity-associated vascular complications.
PVAT function in modulating vascular haemostasis has been extensively reviewed [11][12]. Under normal conditions, the physiological level of ROS is crucial to maintaining vascular homeostasis and is responsible for vascular responses, and excess ROS are antagonised by several antioxidant enzyme systems in PVAT, as mentioned above [30][31]. Pro-inflammatory and pro-oxidative states in PVAT significantly altered the anti-contractile effects and functions of PVAT under obese conditions [32]. For example, during obesity, H2O2 might act as a PVAT-derived contractile factor [27]. PVAT dysfunction leads to the imbalance of PVAT-derived vasoactive factors and affects vascular function [33].
In obesity, the mass of PVAT is increased and adipocytes become hypertrophy, resulting in a shift to white adipose tissue-like characteristics of PVAT, accompanied by deformed mitochondria [29]. Chronic inflammation is evident in obese PVAT, characterised by the infiltration of dendritic cells and macrophage and the upregulation of inflammatory cytokines, including monocyte chemoattractant protein-1 (MCP-1), tumour necrosis factor alpha (TNF-α), IL-6, and the adipokine leptin [34][35]. On the other hand, the expression of adiponectin, an anti-inflammatory adipokine, is reduced in obese PVAT [36]. Inflammation in PVAT also stimulates the generation of O2− and H2O2 by NOX, which promotes the pro-contractile activity of the vessel wall. Also, hypertrophic adipocytes may exhibit insufficient blood perfusion, which leads to local hypoxia in PVAT. The expression of the key modulator of hypoxia, hypoxia-inducible factor alpha (HIF-1α), is increased in the adipose tissues of obese subjects [37]. HIF-1α can stimulate the production of inflammatory mediators, such as TNF-α and IL-6, and suppress the expression of adiponectin from PVAT [38]. In the small mesenteric arteries of healthy Wistar rats, incubation with TNF-α and IL-6 led to the loss of anti-contractile effects of mesenteric PVAT, whereas the induction of hypoxia led to inflammation and dysfunction of mesenteric PVAT [17]. This hypoxia-induced mesenteric PVAT dysfunction was restored by treatment with either IL-6 antibody, TNF-α antibody, or exogenous catalase and SOD in vitro [17]. HFD-induced obese mice with TNF-α receptor knockdown had reduced H2O2 generation in PVAT and sensitivity to phenylephrine(PE)-induced vasocontraction, suggesting that oxidative stress is crucial to the pro-contractile shift of PVAT [27]. In addition, the combination of inflammation and oxidative stress may create a vicious cycle that further generates genetic and cardio-metabolic factors, leading to atherogenesis [39]. Therefore, oxidative stress in PVAT is a critical link between metabolic diseases and cardiovascular complications.
Various studies have also demonstrated that obesity-induced PVAT dysfunction is associated with increased ROS generation from different sources [4][26][27][35]. ob/ob mice showed low activity of GPX and the upregulation of gamma-glutamylcysteine synthetase (γ-GCS), resulting in high glutathione content in adipose tissues [40]. In a study, the expression of SOD2 was significantly reduced and catalase expression was increased in the PVAT from obese mice. Interestingly, the SOD activity was increased, while there was no change in the catalase activity in PVAT. These data suggest a compensatory mechanism for increased ROS in obese PVAT [27]. The thoracic PVAT of obese mice lost its anti-contractile effect and became dysfunctional, which was associated with increased levels of O2− and H2O2 detect by DHE, Amplex red, and lucigenin [27]. An excess of mitochondria-derived ROS may be contribution by the oxidative stress in thoracic PVAT, as evidenced by a significant reduction in the O2 consumption rate and the downregulation of UCP-1 and SOD2 in this tissue [27]. In addition to mitochondrial ROS, eNOS uncoupling also contributes to oxidative stress in thoracic PVAT. In the thoracic PVAT of obese mice, increased arginase activity was detected, which resulted in eNOS uncoupling, while L-arginine supplementation and arginase inhibition reversed the eNOS uncoupling [26]. In patients who underwent bariatric surgery, obese-induced PVAT dysfunction was restored by increased NO production and reduced TNF-α expression [41]. Moreover, thoracic PVAT-conditioned media from obese mice induced H2O2 production in the aortas isolated from control mice in vitro [34], suggesting that the secretome from obese PVAT could be pro-oxidant. The abdominal aortic PVAT of HFD-fed mice exhibited increased mass, adipocyte hypertrophy, and increased levels of O2− and H2O2 (evaluated by luminol chemiluminescence technique) compared to NCD-fed mice [4]. The abdominal PVAT from HFD-fed mice was dysfunctional and the abdominal aorta had impaired endothelium-dependent vasodilation in the presence of obese abdominal PVAT. NOX has been suggested as a source of ROS in obese abdominal aortic PVAT, which was evidenced by the upregulation of p67phox subunit [4]. In long-term HFD-fed rats, increased expressions of cytochrome c oxidase, GPx, and UCP-1 and a decreased expression of p22phox were detected in the aortic PVAT [42]. In the early stages of obesity, the overproduction of NO could preserve vascular function in mesenteric arteries [28]. However, in long-term HFD-induced obesity, mesenteric PVAT became dysfunctional and prooxidant, which was associated with increased O2− production, increased NOX activity, and reduced SOD activity [35]. HFD-fed mice also showed a reduced expression of SOD3 and glutathione levels in mesenteric PVAT [35]. The dysfunction of mesenteric PVAT in long-term HFD-induced obese mice was attenuated by incubation with exogenous sources of SOD and catalase, suggesting the generation of O2− and H2O2 in these dysfunctional mesenteric PVAT [43]. In addition, proteomic analysis of PVAT from gluteal fat biopsy revealed a downregulation of SOD1 and PRX-1 expression in obese individuals [43].
eNOS in PVAT plays an important role in obesity-induced vascular dysfunction [7][11], and researchers have recently reviewed the detailed function of eNOS in PVAT both physiological and pathological conditions [12]. Various studies using HFD and/or genetically modified rodent models have demonstrated the pathophysiological role of eNOS expressed in PVAT in modulating vascular tone, function, and homeostasis, inflammation, and oxidative stress [26][44][45]. Researchers have previously shown evidence of PVAT eNOS dysfunction and eNOS uncoupling in mice with HFD-induced obesity [26]. At the early phase of HFD feeding, there was adaptive NO overproduction from mesenteric PVAT in C57BL/6J mice [28], while the expression of eNOS was reduced after long-term HFD feeding in the mesenteric PVAT of obese rats [46] and in the thoracic PVAT of obese mice [35]. The basal production of NO was reduced in the small arteries of obese patients compared to non-obese subjects, while this reduction was only evident in PVAT-adhered and not in PVAT-removed arteries [47]. The upregulation of arginase in obese PVAT reduces the bioavailability of L-arginine for NO production and leads to the uncoupling of eNOS [48], which in turn produces O2− and increases oxidative stress in PVAT [26].
Macrophages represent the key modulators of oxidative stress and inflammation in PVAT. The upregulation of IL-6 and MCP-1 levels lead to the recruitment of monocytes and macrophage in PVAT and the subsequent pathology of obesity-induced vascular complications [49][50][51]. Also, a reduced adiponectin level in obese PVAT was associated with increased macrophage infiltration [52]. In obese individuals, mineralocorticoid receptors (MR) are activated and their ligand aldosterone is significantly increased [53]. Aldosterone is known to activate NOX [54] and induce eNOS uncoupling [55]. The upregulation of MR increased H2O2 generation in adipocytes in vitro [56] and a blockade of MR prevented both mitochondrial and PVAT dysfunction in obesity [57]. MR may participate in PVAT dysfunction through the modulation of mitochondrial function [57]. Also, MR activation in PVAT macrophages may play a critical role in the pathogenesis of obesity-induced vascular dysfunction, as demonstrated by the beneficial effects in myeloid MR KO mice [58]. Therefore, MR activation is especially interesting in the context of obesity-related cardiovascular and metabolic diseases.
The aldoketo reductase super-family catalyses the generation of sorbitol in the polyol metabolic pathway of glucose metabolism. Aldose reductase, a member of the super-family, may deplete the antioxidant glutathione system due to the scavenging of NADPH, which in turn increases ROS production [59]. In a rat model of type 2 diabetes, the aortic PVAT exhibited increased levels of markers of oxidative stress, including malonaldehyde and aldose reductase activity, which were associated with reduced antioxidant defence [51].
Angiotensin II (Ang II) is the key component of the renin–angiotensin–aldosterone system (RAAS), which has been extensively studied in vascular biology. Ang II mediates the PVAT-associated contractile response to perivascular neuronal excitation [60], while adipocyte RAAS is involved in adipogenesis and adipose tissue mass [61]. The upregulation of Ang II during obesity may lead to adipose tissue dysfunction and induce ROS production in PVAT [62]. In a rat model of heart failure, oxidative stress (measured by DHE fluorescence) and reduced NO bioavailability have been shown to be associated with the impaired anti-contractile effect of thoracic PVAT [63]. In a recent RNA sequencing study, the responses of different PVAT to Ang II have been investigated [64]. Upon stimulation by Ang II, abdominal aortic PVAT showed a significant downregulation of mitochondrial genes in oxidative phosphorylation and brown adipocyte markers and an upregulation of inflammatory markers. In addition, Ang II induced even more significant inflammation in both ascending and descending thoracic aortic PVAT [64]. Together, these targets may emerge as possible mediators of oxidative stress in PVAT during obesity, and further studies are warranted to elucidate the mechanisms (Figure 1).
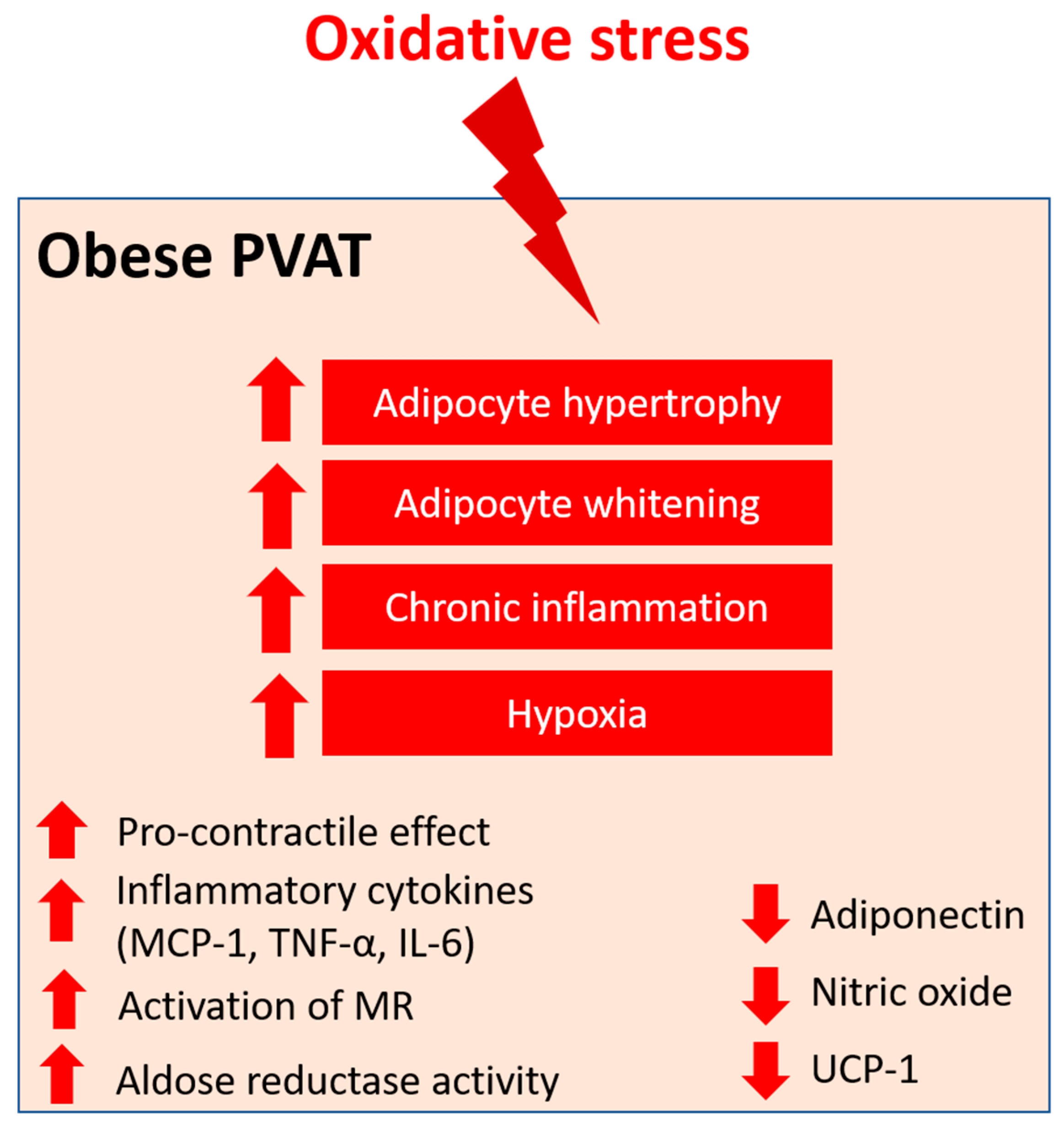
Figure 1. PVAT oxidative stress in obesity. Pro-oxidative state in PVAT significantly alters the anti-contractile effects and functions of PVAT under obese conditions. Obese PVAT becomes hypertrophy and increases whitening of adipocytes. Chronic inflammation and hypoxia are also hallmarks of obese PVAT. In obese PVAT, the anti-contractile effect is lost and becomes pro-contractile. Increased inflammatory cytokines, including MCP-1, TNF-α, and IL-6, are released from PVAT. Also, activation of mineralocorticoid receptors (MR) and increased activity of aldose reductase are recently reported in obese PVAT. The downregulation of UCP-1 in obese PVAT is associated with reduced mitochondrial biogenesis. Obese PVAT also produces less vasoprotective substances like adiponectin and nitric oxide.
3. Pharmacological Prevention of PVAT Oxidative Stress
3.1. Improving Antioxidant Defence
In general, the dismutation of mitochondrial H2O2, the inactivation of O2−, and the uncoupling of oxidative phosphorylation have been demonstrated to restore PVAT function and attenuate PE-induced contraction in vessels with PVAT isolated from HFD-fed mice [4][27]. In HFD-induced obese rats, the administration of antioxidative ethanolic extract of Mangosteen pericarp (EEMP), which contains xanthone, has been shown to normalise hypertrophic PVAT and reduce the expression of vascular cell adhesion molecule 1 (VCAM-1) to prevent arterial remodelling [65]. Treatment with an antioxidant, N-acetyl cysteine, normalised the upregulation of angiotensinogen in ROS-treated adipose tissues in both in vitro culture and in vivo obese mice models [66].
Treatment with either enalaprilat (an angiotensin-converting enzyme ACE inhibitor) or candesartan (an Ang II type 1 receptor antagonist) reduced the PVAT-mediated O2−-induced vasocontraction in rat mesenteric arteries [60]. Also, chronic treatment with quinapril (an ACE inhibitor) reduced the blood pressure and alleviated the potentiation effect of PVAT-mediated superoxide-induced contractions [60]. S-zofenopril, a sulphhydrylated ACE inhibitor, improved vascular function in spontaneous hypertensive rats, which was associated with the potentiation of the H2S pathway [67]. The administration of exogenous H2S inhibited the generation of ROS and suppressed vascular oxidative stress in hypertensive rats [68]. The antioxidant effect of H2S may be attributed to the inhibition of Ang II receptor type 1, the downregulation of NOX, and the upregulation of antioxidant enzymes [69]. Atorvastatin decreases the level of coenzyme Q10, which is a cofactor of H2S oxidation, leading to increased H2S levels. Atorvastatin treatment has been demonstrated to improve the anti-contractile function of PVAT in spontaneously hypertensive rats [70], while the administration of lipophilic atorvastatin increased H2S levels in PVAT and prevented mitochondrial oxidation, which in turn improved the anti-contractile effect of PVAT [71].
Melatonin (5-methoxy-N-acetyltryptamine) is a hormone that has antioxidant activity by promoting direct free radical scavenging and the stimulation of antioxidant enzymes such as SOD [72]. In mice models of accelerated aging, long-term treatment with melatonin normalised the anti-contractile effects of PVAT and was associated with the increased expressions of vasoprotective markers and decreased oxidative stress and inflammation in PVAT [73]. In a recent study, the administration of melatonin restored the anticontractile effect of aortic PVAT in obese rats by reversing the overproduction of ROS, reduced SOD activity, and the decreased bioavailability of NO [74].
Polysaccharide peptides (PsPs) are protein-bound polysaccharide extracted from plants and fungi. The anti-inflammatory, free radical scavenging, and antioxidant properties of PsPs have been demonstrated in different studies [75]. Various studies have shown that PsPs isolated from fungi can restore H2O2 level by upregulating SOD and catalase expression in the PVAT of HFD-fed rats, which in turn prevents PVAT hypertrophy and arterial remodelling [76][77].
Glucagon-like petide-1 (GLP-1) is a peptide that is mainly produced by the intestinal cells and is known to improve cardiovascular health [78], improve endothelial function in obesity [79], and stimulate fatty acid oxidation and insulin signalling pathways, thus enhancing the antioxidant capacity [80]. An antioxidative GLP-1 analogue, liraglutide, has been demonstrated to attenuate HFD-induced vascular dysfunction by modulating the protein kinase A (PKA)-AMP-activated protein kinase (AMPK)-peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1α) pathway in obese mice [81]. Liraglutide enhanced the HO-1/adiponectin axis and alleviated HFD-induced oxidative stress in PVAT [81]. Similar findings were reported in another study where liraglutide increased the antioxidant capacity by upregulating the Nrf2/HO-1 pathway in obese mice [81], and alleviated the NLR family pyrin domain containing 3 (NLRP3) inflammasome-dependent inflammation in PVAT by inhibiting nuclear factor (NF)-κB signalling [82]. Exendin-4, another GLP-1 analogue, reduced the expressions of inflammatory and oxidative markers (such as NOX4) in in vitro and in vivo experiments [83]. On the other hand, dipeptidyl peptidase 4 (DPP-4), an enzyme secreted from PVAT, degrades GLP-1 and has been suggested as a pathophysiological link between obesity and cardiovascular diseases [84]. DPP-4 inhibitors have been shown to exert direct antioxidant effects in rodent models [85][86]. The administration of teneligliptin, a DPP-4 inhibitor, attenuated atherosclerosis progression in apolipoprotein E (ApoE) knockout mice by alleviating inflammation and oxidative stress in both the vasculature and PVAT [87]. These studies suggest that enhancing GLP-1 activity and/or downregulating DPP-4 in PVAT may improve PVAT function by alleviating inflammation and oxidative stress.
3.2. Restoring eNOS Function
PVAT dysfunction can be rescued by restoring the normal expression and function of eNOS. In mice lacking low-density lipoprotein receptors (Ldlrs), thoracic PVAT exhibited compensatorily increased eNOS expression and NO production, which protected against impaired vasodilatation responses to acetylcholine and insulin [88]. Standardised Crataegus extract WS® 1442 is a dry extract from hawthorn leaves with flowers with antioxidative properties [89]. The lab has previously demonstrated that WS® 1442 treatment can restore the vascular function in PVAT-attached aorta rings isolated from HFD-induced obese mice, partly by reversing the reduced Akt (protein kinase B) phosphorylation, reduced eNOS phosphorylation, and enhanced eNOS acetylation in PVAT [90].
The plasma levels of adiponectin and adiponectin expression in adipose tissues are significantly diminished in eNOS global knockout mice [91]. Long-term adiponectin treatment in HFD-fed rats normalised NO-dependent vasorelaxation, which was associated with decreased PVAT inflammation and enhanced eNOS phosphorylation [92]. In a recent study, treatment with methotrexate, an anti-inflammatory drug with antioxidant effects, rescued endothelial and PVAT dysfunction and adipokine dysregulation via activating the AMPK/eNOS pathway in PVAT [93]. Also, treatments with various modulators of AMPK activity, including 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR), diosgenin, metformin, methotrexate, resveratrol, and salicylate, have been shown to increase the anticontractile function of PVAT in different studies [94][95]. In addition, irisin, a newly identified hormone secreted by myocytes, has been shown to attenuate PVAT dysfunction in HFD-induced obese mice via the upregulation of the HO-1/adiponectin axis and browning of the PVAT [96]. In another study, irisin improved endothelial function in obese subjects via activation of the AMPK-eNOS pathway [97], suggesting that the administration of irisin may improve PVAT function by activation of the AMPK-eNOS pathway in PVAT.
Moreover, the expression of eNOS was revealed in both BAT and isolated brown adipocytes [98], whereas eNOS-derived NO has been shown to promote adiponectin synthesis and play a crucial role in mitochondrial biogenesis [99]. These results suggest that restoring eNOS function may also facilitate thermogenesis and browning in PVAT.
References
- GBD 2015 Obesity Collaborators. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 2017, 377, 13–27.
- Lavie, C.J.; Milani, R.V.; Ventura, H.O. Obesity and cardiovascular disease: Risk factor, paradox, and impact of weight loss. J. Am. Coll. Cardiol. 2009, 53, 1925–1932.
- Soltis, E.E.; Cassis, L.A. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin. Exp. Hypertens. Part A Theory Pract. 1991, 13, 277–296.
- Ketonen, J.; Shi, J.; Martonen, E.; Mervaala, E. Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese C57Bl/6 mice. Circ. J. 2010, 74, 1479–1487.
- Qi, X.-Y.; Qu, S.-L.; Xiong, W.-H.; Rom, O.; Chang, L.; Jiang, Z.-S. Perivascular adipose tissue (PVAT) in atherosclerosis: A double-edged sword. Cardiovasc. Diabetol. 2018, 17, 134.
- Zou, L.; Wang, W.; Liu, S.; Zhao, X.; Lyv, Y.; Du, C.; Su, X.; Geng, B.; Xu, G. Spontaneous hypertension occurs with adipose tissue dysfunction in perilipin-1 null mice. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 182–191.
- Xia, N.; Li, H. The role of perivascular adipose tissue in obesity-induced vascular dysfunction. Br. J. Pharmacol. 2017, 174, 3425–3442.
- Fitzgibbons, T.P.; Kogan, S.; Aouadi, M.; Hendricks, G.M.; Straubhaar, J.; Czech, M.P. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am. J. Physiol.-Heart Circ. Physiol. 2011, 301, H1425–H1437.
- Gil-Ortega, M.; Somoza, B.; Huang, Y.; Gollasch, M.; Fernández-Alfonso, M.S. Regional differences in perivascular adipose tissue impacting vascular homeostasis. Trends Endocrinol. Metab. 2015, 26, 367–375.
- Brown, N.K.; Zhou, Z.; Zhang, J.; Zeng, R.; Wu, J.; Eitzman, D.T.; Chen, Y.E.; Chang, L. Perivascular adipose tissue in vascular function and disease: A review of current research and animal models. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1621–1630.
- Man, A.W.C.; Zhou, Y.; Xia, N.; Li, H. Perivascular Adipose Tissue as a Target for Antioxidant Therapy for Cardiovascular Complications. Antioxidants 2020, 9, 574.
- Man, A.W.C.; Zhou, Y.; Xia, N.; Li, H. Endothelial Nitric Oxide Synthase in the Perivascular Adipose Tissue. Biomedicines 2022, 10, 1754.
- Saxton, S.N.; Clark, B.J.; Withers, S.B.; Eringa, E.C.; Heagerty, A.M. Mechanistic Links Between Obesity, Diabetes, and Blood Pressure: Role of Perivascular Adipose Tissue. Physiol. Rev. 2019, 99, 1701–1763.
- Drosos, I.; Chalikias, G.; Pavlaki, M.; Kareli, D.; Epitropou, G.; Bougioukas, G.; Mikroulis, D.; Konstantinou, F.; Giatromanolaki, A.; Ritis, K.; et al. Differences between perivascular adipose tissue surrounding the heart and the internal mammary artery: Possible role for the leptin-inflammation-fibrosis-hypoxia axis. Clin. Res. Cardiol. 2016, 105, 887–900.
- Barp, C.G.; Benedet, P.O.; Assreuy, J. Perivascular adipose tissue phenotype and sepsis vascular dysfunction: Differential contribution of NO, ROS and beta 3-adrenergic receptor. Life Sci. 2020, 254, 117819.
- Gao, Y.J.; Zeng, Z.H.; Teoh, K.; Sharma, A.M.; Abouzahr, L.; Cybulsky, I.; Lamy, A.; Semelhago, L.; Lee, R.M. Perivascular adipose tissue modulates vascular function in the human internal thoracic artery. J. Thorac. Cardiovasc. Surg. 2005, 130, 1130–1136.
- Greenstein, A.S.; Khavandi, K.; Withers, S.B.; Sonoyama, K.; Clancy, O.; Jeziorska, M.; Laing, I.; Yates, A.P.; Pemberton, P.W.; Malik, R.A.; et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 2009, 119, 1661–1670.
- Gräbner, R.; Lötzer, K.; Döpping, S.; Hildner, M.; Radke, D.; Beer, M.; Spanbroek, R.; Lippert, B.; Reardon, C.A.; Getz, G.S. Lymphotoxin β receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J. Exp. Med. 2009, 206, 233–248.
- Campbell, K.A.; Lipinski, M.J.; Doran, A.C.; Skaflen, M.D.; Fuster, V.; McNamara, C.A. Lymphocytes and the adventitial immune response in atherosclerosis. Circ. Res. 2012, 110, 889–900.
- Omar, A.; Chatterjee, T.K.; Tang, Y.; Hui, D.Y.; Weintraub, N.L. Proinflammatory phenotype of perivascular adipocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1631–1636.
- Dos Reis Costa, D.E.F.; Silveira, A.L.M.; Campos, G.P.; Nobrega, N.R.C.; de Araujo, N.F.; de Figueiredo Borges, L.; Dos Santos Aggum Capettini, L.; Ferreira, A.V.M.; Bonaventura, D. High-Carbohydrate Diet Enhanced the Anticontractile Effect of Perivascular Adipose Tissue through Activation of Renin-Angiotensin System. Front. Physiol. 2020, 11, 628101.
- Loperena, R.; Harrison, D.G. Oxidative Stress and Hypertensive Diseases. Med. Clin. N. Am. 2017, 101, 169–193.
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619.
- Agha, M.; Agha, R. The rising prevalence of obesity: Part A: Impact on public health. Int. J. Surg. Oncol. 2017, 2, e17.
- Montero, D.; Walther, G.; Perez-Martin, A.; Roche, E.; Vinet, A. Endothelial dysfunction, inflammation, and oxidative stress in obese children and adolescents: Markers and effect of lifestyle intervention. Obes. Rev. 2012, 13, 441–455.
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Munzel, T.; Daiber, A.; Forstermann, U.; et al. Uncoupling of Endothelial Nitric Oxide Synthase in Perivascular Adipose Tissue of Diet-Induced Obese Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 78–85.
- da Costa, R.M.; Fais, R.S.; Dechandt, C.R.P.; Louzada-Junior, P.; Alberici, L.C.; Lobato, N.S.; Tostes, R.C. Increased mitochondrial ROS generation mediates the loss of the anti-contractile effects of perivascular adipose tissue in high-fat diet obese mice. Br. J. Pharmacol. 2017, 174, 3527–3541.
- Gil-Ortega, M.; Stucchi, P.; Guzman-Ruiz, R.; Cano, V.; Arribas, S.; Gonzalez, M.C.; Ruiz-Gayo, M.; Fernandez-Alfonso, M.S.; Somoza, B. Adaptative nitric oxide overproduction in perivascular adipose tissue during early diet-induced obesity. Endocrinology 2010, 151, 3299–3306.
- Ma, L.; Ma, S.; He, H.; Yang, D.; Chen, X.; Luo, Z.; Liu, D.; Zhu, Z. Perivascular fat-mediated vascular dysfunction and remodeling through the AMPK/mTOR pathway in high-fat diet-induced obese rats. Hypertens. Res. 2010, 33, 446–453.
- Gao, Y.J.; Takemori, K.; Su, L.Y.; An, W.S.; Lu, C.; Sharma, A.M.; Lee, R.M. Perivascular adipose tissue promotes vasoconstriction: The role of superoxide anion. Cardiovasc. Res. 2006, 71, 363–373.
- Awata, W.M.C.; Gonzaga, N.A.; Borges, V.F.; Silva, C.B.P.; Tanus-Santos, J.E.; Cunha, F.Q.; Tirapelli, C.R. Perivascular adipose tissue contributes to lethal sepsis-induced vasoplegia in rats. Eur. J. Pharmacol. 2019, 863, 172706.
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32.
- Fernández-Alfonso, M.S.; Gil-Ortega, M.; García-Prieto, C.F.; Aranguez, I.; Ruiz-Gayo, M.; Somoza, B. Mechanisms of perivascular adipose tissue dysfunction in obesity. Int. J. Endocrinol. 2013, 2013, 402053.
- Bailey-Downs, L.C.; Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Sonntag, W.E.; Csiszar, A.; Ungvari, Z. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: A paracrine mechanism contributing to vascular redox dysregulation and inflammation. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2013, 68, 780–792.
- Gil-Ortega, M.; Condezo-Hoyos, L.; Garcia-Prieto, C.F.; Arribas, S.M.; Gonzalez, M.C.; Aranguez, I.; Ruiz-Gayo, M.; Somoza, B.; Fernandez-Alfonso, M.S. Imbalance between Pro and Anti-Oxidant Mechanisms in Perivascular Adipose Tissue Aggravates Long-Term High-Fat Diet-Derived Endothelial Dysfunction. PLoS ONE 2014, 9, e95312.
- Qiu, T.; Li, M.; Tanner, M.A.; Yang, Y.; Sowers, J.R.; Korthuis, R.J.; Hill, M.A. Depletion of dendritic cells in perivascular adipose tissue improves arterial relaxation responses in type 2 diabetic mice. Metabolism 2018, 85, 76–89.
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.-L. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005, 54, 2277–2286.
- Chen, B.; Lam, K.S.; Wang, Y.; Wu, D.; Lam, M.C.; Shen, J.; Wong, L.; Hoo, R.L.; Zhang, J.; Xu, A. Hypoxia dysregulates the production of adiponectin and plasminogen activator inhibitor-1 independent of reactive oxygen species in adipocytes. Biochem. Biophys. Res. Commun. 2006, 341, 549–556.
- Packard, R.R.; Libby, P. Inflammation in atherosclerosis: From vascular biology to biomarker discovery and risk prediction. Clin. Chem. 2008, 54, 24–38.
- Rebolledo, A.; Rebolledo, O.R.; Marra, C.A.; García, M.E.; Palomo, A.R.R.; Rimorini, L.; Gagliardino, J.J. Early alterations in vascular contractility associated to changes in fatty acid composition and oxidative stress markers in perivascular adipose tissue. Cardiovasc. Diabetol. 2010, 9, 65.
- Aghamohammadzadeh, R.; Greenstein, A.S.; Yadav, R.; Jeziorska, M.; Hama, S.; Soltani, F.; Pemberton, P.W.; Ammori, B.; Malik, R.A.; Soran, H.; et al. Effects of bariatric surgery on human small artery function: Evidence for reduction in perivascular adipocyte inflammation, and the restoration of normal anticontractile activity despite persistent obesity. J. Am. Coll. Cardiol. 2013, 62, 128–135.
- Sanchez, C.; Achard, V.; Grino, M.; Tanguy, S. Long term high-fat diet-induced modification of vascular wall and perivascular adipose tissue-mediated oxidative stress: Consequences for endothelium-independent vascular function in rats. Int. J. Clin. Cardiol. 2017, 4, 097.
- Aghamohammadzadeh, R.; Unwin, R.D.; Greenstein, A.S.; Heagerty, A.M. Effects of obesity on perivascular adipose tissue vasorelaxant function: Nitric oxide, inflammation and elevated systemic blood pressure. J. Vasc. Res. 2015, 52, 299–305.
- Horimatsu, T.; Kim, H.W.; Weintraub, N.L. The Role of Perivascular Adipose Tissue in Non-atherosclerotic Vascular Disease. Front. Physiol. 2017, 8, 969.
- Chang, L.; Garcia-Barrio, M.T.; Chen, Y.E. Perivascular Adipose Tissue Regulates Vascular Function by Targeting Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1094–1109.
- Bussey, C.E.; Withers, S.B.; Aldous, R.G.; Edwards, G.; Heagerty, A.M. Obesity-Related Perivascular Adipose Tissue Damage Is Reversed by Sustained Weight Loss in the Rat. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1377–1385.
- Virdis, A.; Duranti, E.; Rossi, C.; Dell’Agnello, U.; Santini, E.; Anselmino, M.; Chiarugi, M.; Taddei, S.; Solini, A. Tumour necrosis factor-alpha participates on the endothelin-1/nitric oxide imbalance in small arteries from obese patients: Role of perivascular adipose tissue. Eur. Heart J. 2015, 36, 784–794.
- Yang, Z.; Ming, X.F. Arginase: The emerging therapeutic target for vascular oxidative stress and inflammation. Front. Immunol. 2013, 4, 149.
- Huang Cao, Z.F.; Stoffel, E.; Cohen, P. Role of perivascular adipose tissue in vascular physiology and pathology. Hypertension 2017, 69, 770–777.
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326.
- Azul, L.; Leandro, A.; Boroumand, P.; Klip, A.; Seiça, R.; Sena, C.M. Increased inflammation, oxidative stress and a reduction in antioxidant defense enzymes in perivascular adipose tissue contribute to vascular dysfunction in type 2 diabetes. Free Radic. Biol. Med. 2020, 146, 264–274.
- Kawanami, D.; Maemura, K.; Takeda, N.; Harada, T.; Nojiri, T.; Imai, Y.; Manabe, I.; Utsunomiya, K.; Nagai, R. Direct reciprocal effects of resistin and adiponectin on vascular endothelial cells: A new insight into adipocytokine–endothelial cell interactions. Biochem. Biophys. Res. Commun. 2004, 314, 415–419.
- Calhoun, D.A.; Sharma, K. The role of aldosteronism in causing obesity-related cardiovascular risk. Cardiol. Clin. 2010, 28, 517–527.
- Hashikabe, Y.; Suzuki, K.; Jojima, T.; Uchida, K.; Hattori, Y. Aldosterone impairs vascular endothelial cell function. J. Cardiovasc. Pharmacol. 2006, 47, 609–613.
- Leopold, J.A.; Dam, A.; Maron, B.A.; Scribner, A.W.; Liao, R.; Handy, D.E.; Stanton, R.C.; Pitt, B.; Loscalzo, J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat. Med. 2007, 13, 189–197.
- Nguyen Dinh Cat, A.; Antunes, T.T.; Callera, G.E.; Sanchez, A.; Tsiropoulou, S.; Dulak-Lis, M.G.; Anagnostopoulou, A.; He, Y.; Montezano, A.C.; Jaisser, F.; et al. Adipocyte-Specific Mineralocorticoid Receptor Overexpression in Mice Is Associated With Metabolic Syndrome and Vascular Dysfunction: Role of Redox-Sensitive PKG-1 and Rho Kinase. Diabetes 2016, 65, 2392–2403.
- Lefranc, C.; Friederich-Persson, M.; Braud, L.; Palacios-Ramirez, R.; Karlsson, S.; Boujardine, N.; Motterlini, R.; Jaisser, F.; Nguyen Dinh Cat, A. MR (Mineralocorticoid Receptor) Induces Adipose Tissue Senescence and Mitochondrial Dysfunction Leading to Vascular Dysfunction in Obesity. Hypertension 2019, 73, 458–468.
- Manrique, C.; Sowers, J.; Aroor, A.; Jia, G.; Habibi, J.; Mortensen, R.M.; Lastra, G. Mineralocorticoid Receptor Activation In Macrophages Mediates High Fat/high Sucrose Induced Vascular Stiffness in Female Mice. Hypertension 2015, 66, A136.
- Srivastava, S.K.; Yadav, U.C.; Reddy, A.B.; Saxena, A.; Tammali, R.; Shoeb, M.; Ansari, N.H.; Bhatnagar, A.; Petrash, M.J.; Srivastava, S. Aldose reductase inhibition suppresses oxidative stress-induced inflammatory disorders. Chem.-Biol. Interact. 2011, 191, 330–338.
- Lu, C.; Su, L.-Y.; Lee, R.M.; Gao, Y.-J. Mechanisms for perivascular adipose tissue-mediated potentiation of vascular contraction to perivascular neuronal stimulation: The role of adipocyte-derived angiotensin II. Eur. J. Pharmacol. 2010, 634, 107–112.
- Thatcher, S.; Yiannikouris, F.; Gupte, M.; Cassis, L. The adipose renin–angiotensin system: Role in cardiovascular disease. Mol. Cell Endocrinol. 2009, 302, 111–117.
- Ramalingam, L.; Menikdiwela, K.; LeMieux, M.; Dufour, J.M.; Kaur, G.; Kalupahana, N.; Moustaid-Moussa, N. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 1106–1114.
- Fontes, M.T.; Paula, S.M.; Lino, C.A.; Senger, N.; Couto, G.K.; Barreto-Chaves, M.L.M.; Mill, J.G.; Rossoni, L.V. Renin-angiotensin system overactivation in perivascular adipose tissue contributes to vascular dysfunction in heart failure. Clin. Sci. 2020, 134, 3195–3211.
- Wang, Z.; Lu, H.; Garcia-Barrio, M.; Guo, Y.; Zhang, J.; Chen, Y.E.; Chang, L. RNA sequencing reveals perivascular adipose tissue plasticity in response to angiotensin II. Pharmacol. Res. 2022, 178, 106183.
- Wihastuti, T.A.; Aini, F.N.; Tjahjono, C.T.; Heriansyah, T. Dietary Ethanolic Extract of Mangosteen pericarp Reduces VCAM-1, Perivascular Adipose Tissue and Aortic Intimal Medial Thickness in Hypercholesterolemic Rat Model. Open Access Maced. J. Med. Sci. 2019, 7, 3158.
- Okada, S.; Kozuka, C.; Masuzaki, H.; Yasue, S.; Ishii-Yonemoto, T.; Tanaka, T.; Yamamoto, Y.; Noguchi, M.; Kusakabe, T.; Tomita, T. Adipose tissue–specific dysregulation of angiotensinogen by oxidative stress in obesity. Metabolism 2010, 59, 1241–1251.
- Bucci, M.; Vellecco, V.; Cantalupo, A.; Brancaleone, V.; Zhou, Z.; Evangelista, S.; Calderone, V.; Papapetropoulos, A.; Cirino, G. Hydrogen sulfide accounts for the peripheral vascular effects of zofenopril independently of ACE inhibition. Cardiovasc. Res. 2014, 102, 138–147.
- Xiao, L.; Dong, J.-H.; Jin, S.; Xue, H.-M.; Guo, Q.; Teng, X.; Wu, Y.-M. Hydrogen sulfide improves endothelial dysfunction via downregulating BMP4/COX-2 pathway in rats with hypertension. Oxidative Med. Cell Longev. 2016, 2016, 8128957.
- Xue, H.; Zhou, S.; Xiao, L.; Guo, Q.; Liu, S.; Wu, Y. Hydrogen sulfide improves the endothelial dysfunction in renovascular hypertensive rats. Physiol. Res. 2015, 64, 663.
- Zeng, Z.-H.; Zhang, Z.-H.; Luo, B.-H.; He, W.-K.; Liang, L.-Y.; He, C.-C.; Su, C.-J. The functional changes of the perivascular adipose tissue in spontaneously hypertensive rats and the effects of atorvastatin therapy. Clin. Exp. Hypertens. 2009, 31, 355–363.
- Wójcicka, G.; Jamroz-Wiśniewska, A.; Atanasova, P.; Chaldakov, G.N.; Chylińska-Kula, B.; Bełtowski, J. Differential effects of statins on endogenous H2S formation in perivascular adipose tissue. Pharmacol. Res. 2011, 63, 68–76.
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278.
- Agabiti-Rosei, C.; Favero, G.; De Ciuceis, C.; Rossini, C.; Porteri, E.; Rodella, L.F.; Franceschetti, L.; Sarkar, A.M.; Agabiti-Rosei, E.; Rizzoni, D. Effect of long-term treatment with melatonin on vascular markers of oxidative stress/inflammation and on the anticontractile activity of perivascular fat in aging mice. Hypertens. Res. 2017, 40, 41–50.
- Gonzaga, N.A.; Awata, W.M.C.; Ficher, S.P.; Assis, V.O.; Alves, J.V.; Tostes, R.C.; Tirapelli, C.R. Melatonin reverses the loss of the anticontractile effect of perivascular adipose tissue in obese rats. J. Pineal Res. 2021, 70, e12710.
- Xu, Z.; Chen, X.; Zhong, Z.; Chen, L.; Wang, Y. Ganoderma lucidum polysaccharides: Immunomodulation and potential anti-tumor activities. Am. J. Chin. Med. 2011, 39, 15–27.
- Wihastuti, T.A.; Sargowo, D.; Heriansyah, T.; Aziza, Y.E.; Puspitarini, D.; Iwana, A.N.; Evitasari, L.A. The reduction of aorta histopathological images through inhibition of reactive oxygen species formation in hypercholesterolemia rattus norvegicus treated with polysaccharide peptide of Ganoderma lucidum. Iran. J. Basic Med. Sci. 2015, 18, 514.
- Prasetya, I.; Wihastuti, T.A.; Widodo, M.A.; Sargowo, D. Ganodermalucidum Polysaccharides Peptide: Possibility of Hypertension Therapy with Antioxidant. J. Hypertens. 2015, 33, e38.
- Standl, E. GLP-1 receptor agonists and cardiovascular outcomes: An updated synthesis. Lancet Diabetes Endocrinol. 2019, 7, 741–743.
- Han, L.; Yu, Y.; Sun, X.; Wang, B. Exendin-4 directly improves endothelial dysfunction in isolated aortas from obese rats through the cAMP or AMPK–eNOS pathways. Diabetes Res. Clin. Pract. 2012, 97, 453–460.
- Mehdi, S.F.; Pusapati, S.; Anwar, M.S.; Lohana, D.; Kumar, P.; Nandula, S.A.; Nawaz, F.K.; Tracey, K.; Yang, H.; LeRoith, D.; et al. Glucagon-like peptide-1: A multi-faceted anti-inflammatory agent. Front. Immunol. 2023, 14, 1148209.
- Han, F.; Hou, N.; Liu, Y.; Huang, N.; Pan, R.; Zhang, X.; Mao, E.; Sun, X. Liraglutide improves vascular dysfunction by regulating a cAMP-independent PKA-AMPK pathway in perivascular adipose tissue in obese mice. Biomed. Pharmacother. 2019, 120, 109537.
- Chen, X.; Huang, Q.; Feng, J.; Xiao, Z.; Zhang, X.; Zhao, L. GLP-1 alleviates NLRP3 inflammasome-dependent inflammation in perivascular adipose tissue by inhibiting the NF-kappaB signalling pathway. J. Int. Med. Res. 2021, 49, 300060521992981.
- Ding, X.; Saxena, N.K.; Lin, S.; Gupta, N.A.; Anania, F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology 2006, 43, 173–181.
- Ussher, J.R.; Drucker, D.J. Cardiovascular biology of the incretin system. Endocr. Rev. 2012, 33, 187–215.
- Alam, M.A.; Chowdhury, M.R.H.; Jain, P.; Sagor, M.A.T.; Reza, H.M. DPP-4 inhibitor sitagliptin prevents inflammation and oxidative stress of heart and kidney in two kidney and one clip (2K1C) rats. Diabetol. Metab. Syndr. 2015, 7, 107.
- Liu, L.; Liu, J.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Xu, A.; Xu, G.; Ng, C.F.; Yao, X.; Gao, Y. Uncoupling protein-2 mediates DPP-4 inhibitor-induced restoration of endothelial function in hypertension through reducing oxidative stress. Antioxid. Redox Signal. 2014, 21, 1571–1581.
- Salim, H.M.; Fukuda, D.; Higashikuni, Y.; Tanaka, K.; Hirata, Y.; Yagi, S.; Soeki, T.; Shimabukuro, M.; Sata, M. Teneligliptin, a dipeptidyl peptidase-4 inhibitor, attenuated pro-inflammatory phenotype of perivascular adipose tissue and inhibited atherogenesis in normoglycemic apolipoprotein-E-deficient mice. Vasc. Pharmacol. 2017, 96, 19–25.
- Baltieri, N.; Guizoni, D.M.; Victorio, J.A.; Davel, A.P. Protective role of perivascular adipose tissue in endothelial dysfunction and insulin-induced vasodilatation of hypercholesterolemic LDL receptor-deficient mice. Front. Physiol. 2018, 9, 229.
- Holubarsch, C.J.F.; Colucci, W.S.; Eha, J. Benefit-Risk Assessment of Crataegus Extract WS 1442: An Evidence-Based Review. Am. J. Cardiovasc. Drugs Drugs Devices Other Interv. 2018, 18, 25–36.
- Xia, N.; Weisenburger, S.; Koch, E.; Burkart, M.; Reifenberg, G.; Förstermann, U.; Li, H. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br. J. Pharmacol. 2017, 174, 3443–3453.
- Koh, E.H.; Kim, M.; Ranjan, K.; Kim, H.S.; Park, H.-S.; Oh, K.S.; Park, I.-S.; Lee, W.J.; Kim, M.-S.; Park, J.-Y. eNOS plays a major role in adiponectin synthesis in adipocytes. Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E846–E853.
- Sena, C.M.; Pereira, A.; Fernandes, R.; Letra, L.; Seica, R.M. Adiponectin improves endothelial function in mesenteric arteries of rats fed a high-fat diet: Role of perivascular adipose tissue. Br. J. Pharmacol. 2017, 174, 3514–3526.
- Ma, Y.; Li, L.; Shao, Y.; Bai, X.; Bai, T.; Huang, X. Methotrexate improves perivascular adipose tissue/endothelial dysfunction via activation of AMPK/eNOS pathway. Mol. Med. Rep. 2017, 15, 2353–2359.
- Chen, Y.; Xu, X.; Zhang, Y.; Liu, K.; Huang, F.; Liu, B.; Kou, J. Diosgenin regulates adipokine expression in perivascular adipose tissue and ameliorates endothelial dysfunction via regulation of AMPK. J. Steroid Biochem. Mol. Biol. 2016, 155, 155–165.
- Sun, Y.; Li, J.; Xiao, N.; Wang, M.; Kou, J.; Qi, L.; Huang, F.; Liu, B.; Liu, K. Pharmacological activation of AMPK ameliorates perivascular adipose/endothelial dysfunction in a manner interdependent on AMPK and SIRT1. Pharmacol. Res. 2014, 89, 19–28.
- Hou, N.; Du, G.; Han, F.; Zhang, J.; Jiao, X.; Sun, X. Irisin Regulates Heme Oxygenase-1/Adiponectin Axis in Perivascular Adipose Tissue and Improves Endothelial Dysfunction in Diet-Induced Obese Mice. Cell Physiol. Biochem. Int. J. Exp. Cell Physiol. Biochem. Pharmacol. 2017, 42, 603–614.
- Han, F.; Zhang, S.; Hou, N.; Wang, D.; Sun, X. Irisin improves endothelial function in obese mice through the AMPK-eNOS pathway. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1501–H1508.
- Kikuchi-Utsumi, K.; Gao, B.; Ohinata, H.; Hashimoto, M.; Yamamoto, N.; Kuroshima, A. Enhanced gene expression of endothelial nitric oxide synthase in brown adipose tissue during cold exposure. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2002, 282, R623–R626.
- Csiszar, A.; Labinskyy, N.; Pinto, J.T.; Ballabh, P.; Zhang, H.; Losonczy, G.; Pearson, K.; De Cabo, R.; Pacher, P.; Zhang, C. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H13–H20.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
980
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
17 Aug 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No