 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Zhenrui PAN | -- | 2479 | 2023-08-10 11:46:48 | | | |
| 2 | Lindsay Dong | + 5 word(s) | 2484 | 2023-08-11 02:57:55 | | | | |
| 3 | Lindsay Dong | Meta information modification | 2484 | 2023-08-11 02:58:24 | | |
Video Upload Options
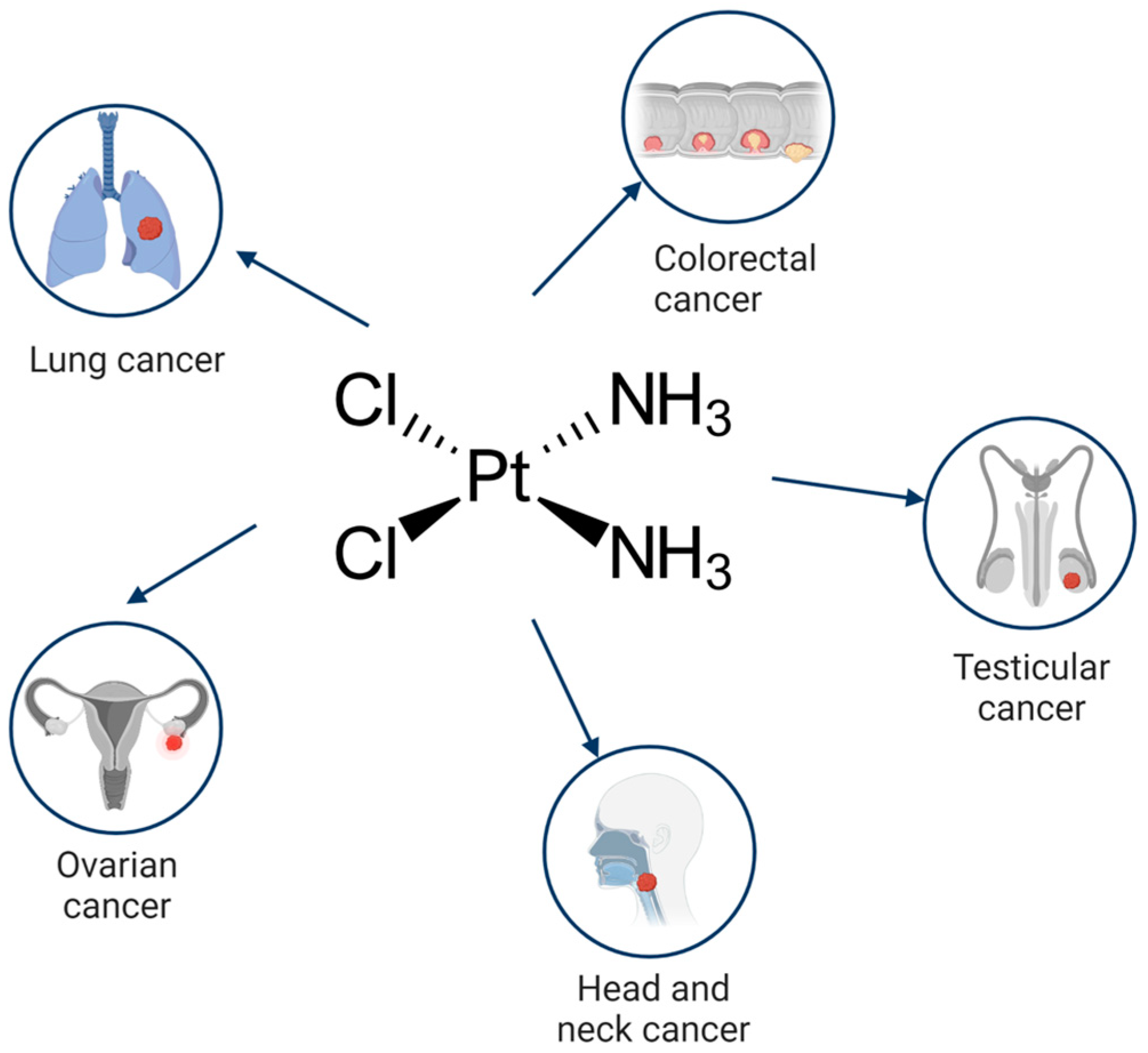
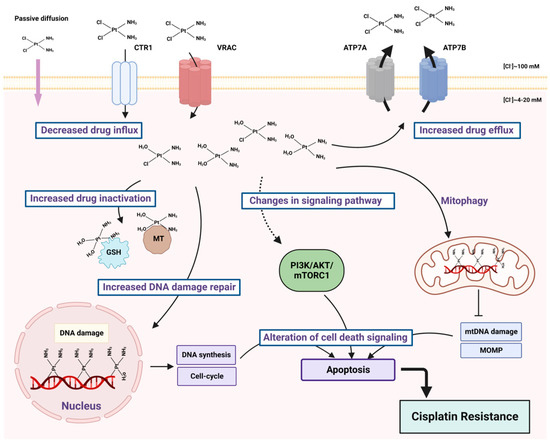
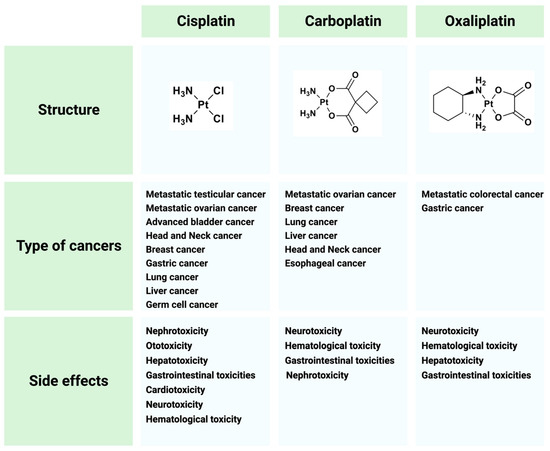
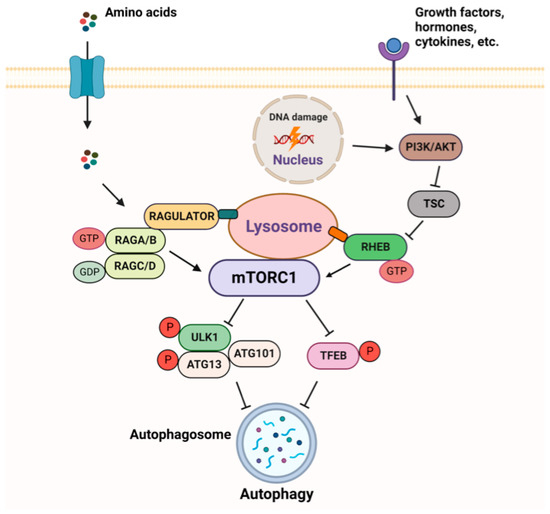
Cisplatin (cis-diamminedichloroplatinum I) is a platinum-based drug, the mainstay of anticancer treatment for numerous solid tumors. Drug resistance is a serious problem in the treatment with platinum-based drugs. Resistance to cisplatin depends on both the inner adaptive mechanisms of cancer cells and the tumor microenvironment, where hypoxic conditions increase the tolerance of cancer cells to the drug. Among intercellular adaptive factors, the most important are: (1) a reduced drug accumulation due to either a decreased influx or an increased efflux; (2) an increase in DNA repair and changes in DNA damage response (DDR); (3) an alteration of apoptosis; (4) changes in signaling pathways, notably the mammalian target of rapamycin complex 1 (mTORC1) pathway.
1. Biochemical Mechanisms of Cisplatin Cytotoxicity
2. Molecular Basis of the mTORC1 Pathway and Autophagy
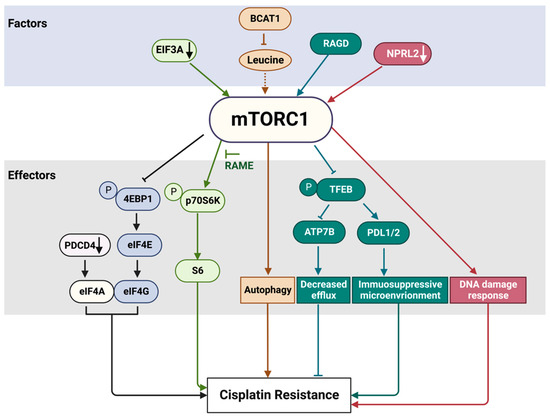
3. mTORC1 Pathway and Cisplatin Resistance
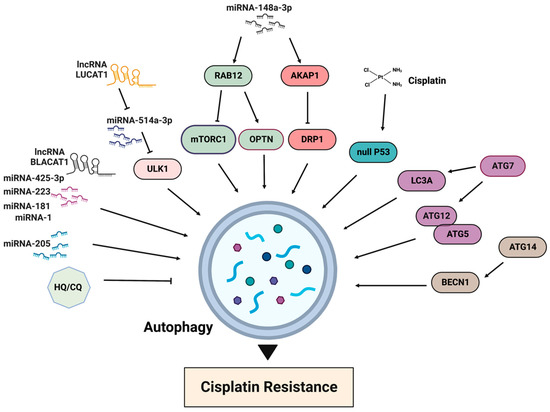
4. Autophagy in Cisplatin Resistance
5. Conclusions
References
- Howell, S.B.; Safaei, R.; Larson, C.A.; Sailor, M.J. Copper Transporters and the Cellular Pharmacology of the Platinum-Containing Cancer Drugs. Mol. Pharmacol. 2010, 77, 887–894.
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit Composition of VRAC Channels Determines Substrate Specificity and Cellular Resistance to P T-based Anti-cancer Drugs. EMBO J. 2015, 34, 2993–3008.
- Lukanović, D.; Herzog, M.; Kobal, B.; Černe, K. The Contribution of Copper Efflux Transporters ATP7A and ATP7B to Chemoresistance and Personalized Medicine in Ovarian Cancer. Biomed. Pharmacother. 2020, 129, 110401.
- Petruzzelli, R.; Polishchuk, R.S. Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs. Cells 2019, 8, 1080.
- Jamieson, E.R.; Lippard, S.J. Structure, Recognition, and Processing of Cisplatin−DNA Adducts. Chem. Rev. 1999, 99, 2467–2498.
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists. Dalton Trans. 2018, 47, 6645–6653.
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorgan. Chem. 2019, 88, 102925.
- Ranasinghe, R.; Mathai, M.L.; Zulli, A. Cisplatin for Cancer Therapy and Overcoming Chemoresistance. Heliyon 2022, 8, e10608.
- Battaglioni, S.; Benjamin, D.; Wälchli, M.; Maier, T.; Hall, M.N. MTOR Substrate Phosphorylation in Growth Control. Cell 2022, 185, 1814–1836.
- Ben-Sahra, I.; Manning, B.D. MTORC1 Signaling and the Metabolic Control of Cell Growth. Curr. Opin. Cell Biol. 2017, 45, 72–82.
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203.
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to MTORC1. Science 2008, 320, 1496–1501.
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in Nutrient Response. Nat. Cell Biol. 2008, 10, 935–945.
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator Is a GEF for the Rag GTPases That Signal Amino Acid Levels to MTORC1. Cell 2012, 150, 1196–1208.
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. MTORC1 Senses Lysosomal Amino Acids through an Inside-out Mechanism That Requires the Vacuolar H+-ATPase. Science 2011, 334, 678–683.
- Long, X.; Ortiz-Vega, S.; Lin, Y.; Avruch, J. Rheb Binding to Mammalian Target of Rapamycin (MTOR) Is Regulated by Amino Acid Sufficiency. J. Biol. Chem. 2005, 280, 23433–23436.
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat. Rev. Cancer 2020, 20, 74–88.
- Hu, Y.; Reggiori, F. Molecular Regulation of Autophagosome Formation. Biochem. Soc. Trans. 2022, 50, 55–69.
- Du Rusquec, P.; Blonz, C.; Frenel, J.S.; Campone, M. Targeting the PI3K/Akt/MTOR Pathway in Estrogen-Receptor Positive HER2 Negative Advanced Breast Cancer. Ther. Adv. Med. Oncol. 2020, 12, 175883592094093.
- Li, H.; Zeng, J.; Shen, K. PI3K/AKT/MTOR Signaling Pathway as a Therapeutic Target for Ovarian Cancer. Arch. Gynecol. Obstet. 2014, 290, 1067–1078.
- Marquard, F.E.; Jücker, M. PI3K/AKT/MTOR Signaling as a Molecular Target in Head and Neck Cancer. Biochem. Pharmacol. 2020, 172, 113729.
- Tan, A.C. Targeting the PI3K/Akt/MTOR Pathway in Non-small Cell Lung Cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518.
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/MTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128.
- Gremke, N.; Polo, P.; Dort, A.; Schneikert, J.; Elmshäuser, S.; Brehm, C.; Klingmüller, U.; Schmitt, A.; Reinhardt, H.C.; Timofeev, O.; et al. MTOR-Mediated Cancer Drug Resistance Suppresses Autophagy and Generates a Druggable Metabolic Vulnerability. Nat. Commun. 2020, 11, 4684.
- Zhang, X.; Wang, X.; Song, X.; Liu, C.; Shi, Y.; Wang, Y.; Afonja, O.; Ma, C.; Chen, Y.H.; Zhang, L. Programmed Cell Death 4 Enhances Chemosensitivity of Ovarian Cancer Cells by Activating Death Receptor Pathway in Vitro and in Vivo. Cancer Sci. 2010, 101, 2163–2170.
- Liu, R.-Y.; Dong, Z.; Liu, J.; Yin, J.-Y.; Zhou, L.; Wu, X.; Yang, Y.; Mo, W.; Huang, W.; Khoo, S.K.; et al. Role of EIF3a in Regulating Cisplatin Sensitivity and in Translational Control of Nucleotide Excision Repair of Nasopharyngeal Carcinoma. Oncogene 2011, 30, 4814–4823.
- Yin, J.-Y.; Shen, J.; Dong, Z.-Z.; Huang, Q.; Zhong, M.-Z.; Feng, D.-Y.; Zhou, H.-H.; Zhang, J.-T.; Liu, Z.-Q. Effect of EIF3a on Response of Lung Cancer Patients to Platinum-Based Chemotherapy by Regulating DNA Repair. Clin. Cancer Res. 2011, 17, 4600–4609.
- Hsu, Y.-C.; Chern, J.J.; Cai, Y.; Liu, M.; Choi, K.-W. Drosophila TCTP Is Essential for Growth and Proliferation through Regulation of DRheb GTPase. Nature 2007, 445, 785–788.
- Bommer, U.-A.; Telerman, A. Dysregulation of TCTP in Biological Processes and Diseases. Cells 2020, 9, 1632.
- Jeong, M.; Jeong, M.H.; Kim, J.E.; Cho, S.; Lee, K.J.; Park, S.; Sohn, J.; Park, Y.G. TCTP Protein Degradation by Targeting MTORC1 and Signaling through S6K, Akt, and Plk1 Sensitizes Lung Cancer Cells to DNA-Damaging Drugs. Sci. Rep. 2021, 11, 20812.
- Jiang, W.; Ou, Z.; Zhu, Q.; Zai, H. RagC GTPase Regulates MTOR to Promote Chemoresistance in Senescence-like HepG2 Cells. Front. Physiol. 2022, 13, 949737.
- Loissell-Baltazar, Y.A.; Dokudovskaya, S. SEA and GATOR 10 Years Later. Cells 2021, 10, 2689.
- Ueda, K.; Kawashima, H.; Ohtani, S.; Deng, W.-G.G.; Ravoori, M.; Bankson, J.; Gao, B.; Girard, L.; Minna, J.D.; Roth, J.A.; et al. The 3p21.3 Tumor Suppressor NPRL2 Plays an Important Role in Cisplatin-Induced Resistance in Human Non-Small-Cell Lung Cancer Cells. Cancer Res. 2006, 66, 9682–9690.
- Jayachandran, G.; Ueda, K.; Wang, B.; Roth, J.A.; Ji, L. NPRL2 Sensitizes Human Non-Small Cell Lung Cancer (NSCLC) Cells to Cisplatin Treatment by Regulating Key Components in the DNA Repair Pathway. PLoS ONE 2010, 5, e11994.
- Ma, Y.; Silveri, L.; LaCava, J.; Dokudovskaya, S. Tumor Suppressor NPRL2 Induces ROS Production and DNA Damage Response. Sci. Rep. 2017, 7, 15311.
- Ren, J.-H.; He, W.-S.; Nong, L.; Zhu, Q.-Y.; Hu, K.; Zhang, R.-G.; Huang, L.-L.; Zhu, F.; Wu, G. Acquired Cisplatin Resistance in Human Lung Adenocarcinoma Cells Is Associated with Enhanced Autophagy. Cancer Biother. Radiopharm. 2010, 25, 75–80.
- Hwang, J.R.; Kim, W.Y.; Cho, Y.-J.; Ryu, J.-Y.; Choi, J.-J.; Jeong, S.Y.; Kim, M.-S.; Kim, J.H.; Paik, E.S.; Lee, Y.-Y.; et al. Chloroquine Reverses Chemoresistance via Upregulation of P21WAF1/CIP1 and Autophagy Inhibition in Ovarian Cancer. Cell Death Dis. 2020, 11, 1034.
- Huang, D.; Savage, S.R.; Calinawan, A.P.; Lin, C.; Zhang, B.; Wang, P.; Starr, T.K.; Birrer, M.J.; Paulovich, A.G. A Highly Annotated Database of Genes Associated with Platinum Resistance in Cancer. Oncogene 2021, 40, 6395–6405.
- Miyamoto, M.; Takano, M.; Aoyama, T.; Soyama, H.; Yoshikawa, T.; Tsuda, H.; Furuya, K. Inhibition of Autophagy Protein LC3A as a Therapeutic Target in Ovarian Clear Cell Carcinomas. J. Gynecol. Oncol. 2017, 28, e33.
- Hu, X.; Ma, Z.; Wen, L.; Li, S.; Dong, Z. Autophagy in Cisplatin Nephrotoxicity during Cancer Therapy. Cancers 2021, 13, 5618.
- Yu, G.; Klionsky, D.J. Life and Death Decisions—The Many Faces of Autophagy in Cell Survival and Cell Death. Biomolecules 2022, 12, 866.
- Zhao, J.; Nie, Y.; Wang, H.; Lin, Y. MiR-181a Suppresses Autophagy and Sensitizes Gastric Cancer Cells to Cisplatin. Gene 2016, 576, 828–833.
- Li, B.; Wang, W.; Li, Z.; Chen, Z.; Zhi, X.; Xu, J.; Li, Q.; Wang, L.; Huang, X.; Wang, L.; et al. MicroRNA-148a-3p Enhances Cisplatin Cytotoxicity in Gastric Cancer through Mitochondrial Fission Induction and Cyto-Protective Autophagy Suppression. Cancer Lett. 2017, 410, 212–227.
- Matsui, T.; Fukuda, M. Rab12 Regulates MTORC1 Activity and Autophagy through Controlling the Degradation of Amino-acid Transporter PAT4. EMBO Rep. 2013, 14, 450–457.
- Sirohi, K.; Chalasani, M.L.S.; Sudhakar, C.; Kumari, A.; Radha, V.; Swarup, G. M98K-OPTN Induces Transferrin Receptor Degradation and RAB12—Mediated Autophagic Death in Retinal Ganglion Cells. Autophagy 2013, 9, 510–527.
- Zou, G.-P.; Yu, C.-X.; Shi, S.-L.; Li, Q.-G.; Wang, X.-H.; Qu, X.-H.; Yang, Z.-J.; Yao, W.-R.; Yan, D.-D.; Jiang, L.-P.; et al. Mitochondrial Dynamics Mediated by DRP1 and MFN2 Contributes to Cisplatin Chemoresistance in Human Ovarian Cancer SKOV3 Cells. J. Cancer 2021, 12, 7358–7373.

