Cisplatin (cis-diamminedichloroplatinum I) is a platinum-based drug, the mainstay of anticancer treatment for numerous solid tumors. Drug resistance is a serious problem in the treatment with platinum-based drugs. Resistance to cisplatin depends on both the inner adaptive mechanisms of cancer cells and the tumor microenvironment, where hypoxic conditions increase the tolerance of cancer cells to the drug. Among intercellular adaptive factors, the most important are: (1) a reduced drug accumulation due to either a decreased influx or an increased efflux; (2) an increase in DNA repair and changes in DNA damage response (DDR); (3) an alteration of apoptosis; (4) changes in signaling pathways, notably the mTORC1 pathway.
1. Biochemical Mechanisms of Cisplatin Cytotoxicity
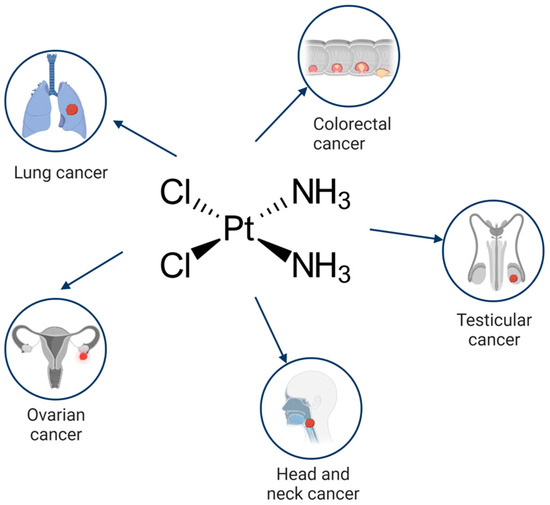
Cisplatin is a first line drug for many cancers, especially for lung, ovarian, head and neck, testicular and colorectal cancers (Figure 1). During anticancer therapy, cisplatin is injected intravenously. A high concentration of chloride ions (~100 mM) in the bloodstream suppresses the hydrolysis of the drug and maintains it in a neutral state. The binding of cisplatin to the plasma proteins, mainly to albumin, results in the deactivation of up to 95% of the injected drug.
Figure 1. Cancers where cisplatin is used as a mainstream drug.
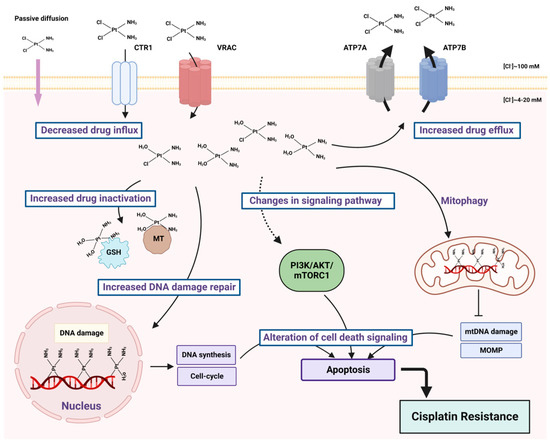
Cisplatin enters the cells by passive diffusion through plasma membrane or by active transport via copper influx transporter 1 (CTR1) [
1] and volume-regulated anion channel (VRAC) [
2] (
Figure 2). The majority of cytoplasmic cisplatin accumulates in vesicles that contain copper efflux transporters ATP7A or ATP7B rather than being diffusely localized throughout the cytoplasm. These intracellular secretory vesicles can further traffic and fuse with the plasma membrane, releasing cisplatin by exocytosis as a free drug, as a conjugate or as a complex with cellular proteins [
3]. Copper transporters can be used by malignant cells to detoxify cisplatin, thereby promoting tumor resistance to chemotherapy [
4].
Figure 2. Main factors of cisplatin resistance. Cancer cells can develop resistance to cisplatin via decreased drug influx or increased efflux, because of the drug inactivation through the interaction with glutathione (GSH) and metallothioneins (MT), because of enhanced DNA damage response and alternations in signaling pathways.
Once in the cytoplasm, cisplatin undergoes aquation due to much lower chloride concentrations (~4–20 mM) than in the bloodstream. The displacement of one or two chloride ions by water molecules results in the generation of a highly electrophilic molecule that can interact with nucleic acids, phospholipids and proteins.
DNA is a primary target of cisplatin. Aquated cisplatin induces DNA damage by the forming of intrastrand and interstrand DNA cross-links through the preferential binding to the N
7 position of guanine [
5]. DNA lesions that are not resolved by DNA repair pathways block the production of DNA, mRNA and proteins and activate several transduction pathways, which finally lead to necrosis or apoptosis. Many cancers have defective DNA repair pathways; therefore, while normal cells can cope with the harm caused by cisplatin, cancer cells will die.
Cisplatin does not show its highest potential in anticancer treatment because of side effects and drug resistance. The major adverse events that arise from cisplatin therapy are nephro-, hepato-, neuro- and gastrointestinal toxicities [
12,
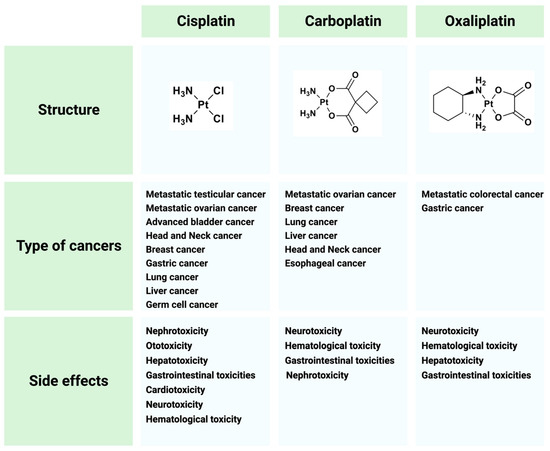
13]. To overcome these limitations, great efforts have been made to search for cisplatin analogues which are better tolerated by patients and/or show anticancer activity in cisplatin-resistant tumors. The most widely used cisplatin derivatives are carboplatin and oxaliplatin (
Figure 3).
Figure 3. Cisplatin and its derivatives used in the treatment of different types of cancers. Side effects for each drug are listed.
Drug resistance is a serious problem in the treatment with platinum-based drugs. Cisplatin resistance could be both intrinsic (occurs from the beginning of treatment) and acquired (initially sensitive cells develop resistance to the drug over time). One of the major determinants of resistance is the type of cancer [
17].
2. Molecular Basis of the mTORC1 Pathway and Autophagy
Mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase that belongs to the phosphatidylinositol 3-kinase PI3K-related family (PIKK). As a part of two structurally and functionally different complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2), mTOR maintains the balance between anabolism and catabolism in response to nutritional or environmental conditions via the phosphorylation of its multiple substrates [
29]. Among nearly 60 direct targets of mTORC1, the most known and well-characterized are p70S6 Kinase 1 (S6K1) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), involved in protein translation; transcription factor EB (TFEB), important for lysosome biogenesis and lipid metabolism; and Unc-51-like autophagy activating kinase 1 (ULK1), a member of the autophagy initiation complex.
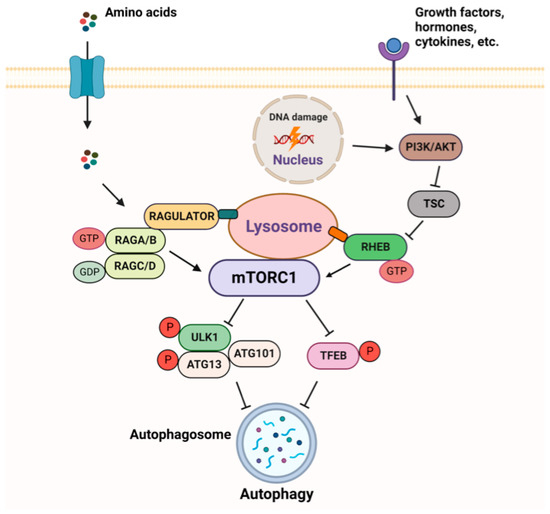
The mTORC1 pathway is subjected to a tight regulation, allowing its activation, when growth factors, energy, and nutrients are sufficient [
30,
31] (
Figure 4). In order to have an adequate and timely response to extra and intracellular inputs, mTORC1 responds to upstream signals through two different sets of the small GTPases–RHEB (Ras homologue enriched in brain) and RAGs (RAG guanosine triphosphatases). The activity of these GTPases depends on their effectors, GTPase-activating proteins (GAPs), which stimulate GTP hydrolysis, and guanine nucleotide exchange factors (GEFs).
Figure 4. Overview of mTORC1 signaling.
The major site of mTORC1 activation is the lysosomal surface, where mTORC1 is recruited and induced in a RAG-GTPase-dependent manner when amino acids are abundant [
32,
33]. Four RAGs exist as heterodimers (i.e., RAGA (or RAGB) with RAGC (or RAGD)). In the presence of amino acids, RAGA/B is loaded with GTP while RAGC/D is bound to GDP. RAGs interact with the pentameric RAGULATOR complex, which exerts a GEF activity towards RAGA or RAGB [
34,
35]. Active RAGULATOR-RAG stimulates the recruitment of mTORC1 to the lysosomal membrane where it is fully activated by RHEB, loaded with GTP [
36]. RHEB is under the control of another signaling node, the tuberous sclerosis (TSC) complex, which acts as a GAP to inhibit RHEB. TSC is a nexus of multiple physiological stimuli (e.g., energy status, growth factors, DNA damage) that signal to mTORC1 through the PI3K-AKT network [
37].
mTORC1 plays a central role as a negative regulator of autophagy, a catabolic process by which cytosol and organelles are sequestered within double-membrane-bound vesicles that deliver their contents to the lysosome for degradation and recycling [
42]. mTORC1 is active and autophagy is suppressed under optimal growth conditions, e.g., a sufficient quantity of amino acids and glucose.
3. mTORC1 Pathway and Cisplatin Resistance
In normal physiological conditions, the PI3K/AKT/mTOR pathway undergoes stringent regulation to ensure the proper activity and balance necessary for healthy homeostasis. In the context of numerous cancers, this pathway exhibits a persistent state of activation. Thus, the aberrant activation of the pathway has been detected in ~70% of ovarian and breast cancers [
46,
47] and in ~90% of head and neck cancers and lung adenocarcinoma [
26,
48]. The mechanisms behind this activation include the amplification of the mutations of genes encoding PI3K subunits, AKT, inactivating mutations in TSC genes, or, conversely, activating mutations in MTOR [
49].
Tumors with acquired resistance to cisplatin often have a constitutive activation of mTORC1 signaling [
53]. mTORC1 can influence cisplatin resistance in many ways—at the transcriptional and translational levels and by responding to various cues, such as amino acids, energy, and DNA damage (
Figure 5).
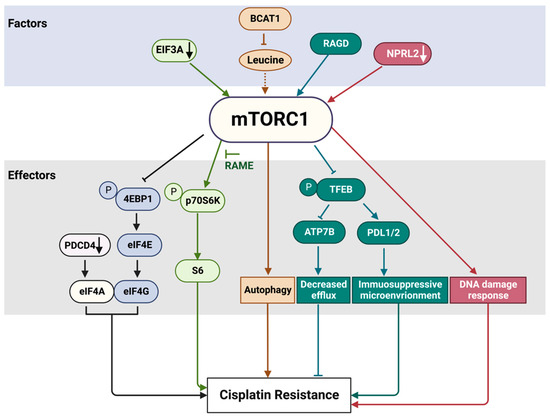
Figure 5. mTORC1 pathway factors and effectors involved in cisplatin resistance. The factors and effectors of the same signaling path are indicated with the same color. EIF3A translational factor negatively regulates mTORC1 activity. Accordingly, EIF3A downregulation (black arrow) increases mTORC1 activity. The activation of p70S6K by mTORC1 can be suppressed by RAME. Many oncogenic mutations have been detected in eukaryotic translation initiation factors, a group of mTORC1 downstream effectors. In addition, PDCD4, which is not a direct target of mTORC1, can suppress protein translation by interacting with EIF4A and EIF4G to inhibit the formation of the translation initiation complex. A knockdown of PDCD4 (black arrow) reduces sensitivity to platinum drugs. BCAT1 overexpression results in a decrease of leucine and other branched-chain amino acid levels. As a consequence, mTORC1 cannot be effectively activated (dashed arrow), resulting in enhanced autophagy and cisplatin resistance. NPRL2 downregulation (white arrow) results in mTORC1 activation, compromised DNA damage response and cisplatin resistance. See text for more details. RAGD promotes TFEB inhibition through its phosphorylation by mTORC1 and sequestration in the cytoplasm, where TFEB cannot exert its function as a transcription factor. Cisplatin treatment induces TFEB nuclear translocation and activation. Active TFEB increases the expression of programmed cell death-ligands 1 and 2 (PD-L1 and PD-L2) to foster an immunosuppressive tumor microenvironment that mediates drug resistance. The suppression of TFEB inhibits the expression of the copper transporter ATP7B involved in cisplatin efflux and sensitizes initially resistant ovarian cancer cells to cisplatin.
Programmed cell death 4 (PDCD4) protein, which is not a direct target of mTORC1, can suppress protein translation by interacting with EIF4A and EIF4G to inhibit the formation of the translation initiation complex. An overexpression of
PDCD4 enhances platinum sensitivity, while a knockdown of PDCD4 reduces platinum sensitivity in ovarian cells and in a xenograft model [
60].
EIF3A, the largest subunit of the eIF3 translational initiation complex, downregulates the translation of a number of nucleotide excision repair proteins. EIF3A knockdown or ectopic overexpression, respectively, increases or decreases cellular resistance to cisplatin in a number of cancer cell lines, including nasopharyngeal and ovarian carcinoma and lung cancer cell lines, likely due to EIF3A’s role in the regulation of NER proteins [
61,
62].
Translationally controlled tumor protein (TCTP) stimulates mTORC1 by positively regulating RHEB activity [
64]. TCTP is overexpressed in many human tumor tissues [
65]. The inhibition of mTORC1 by rapamycin in human lung cancer cells and an A549 lung cancer xenograft model induces ubiquitin–proteasome degradation of TCTP. Moreover, the minimal dose of rapamycin required to induce TCTP proteolysis enhances the efficacy of cisplatin through the induction of apoptotic cell death in vitro and in vivo. This synergistic cytotoxicity was induced irrespective of the functional status of p53 [
66].
Chemotherapy can induce oxidative and genotoxic stress, triggering a senescence-like state, which in many cancer cells causes treatment resistance, supporting tumor proliferation and cancer recurrence. mTORC1 activity is upregulated in senescent cells, which are insensitive to serum and amino acid starvation. How this can be related to the senescence-induced drug resistance was addressed in a recent study by Jiang et al., who investigated the role of five small GTPases that can activate mTORC1 in response to amino acid stimulation [
67]. In the senescence-like hepatoma cell line HepG2, RAGC and RHEB, but not RAB1A, RAB5 or ARF1, were required for persistent mTORC1 activity.
One of the major upstream regulators of the mTORC1 pathway, the GATOR1 complex, is involved in the regulation of nutrient sensing and responding. Various mutations of the genes encoding GATOR1 proteins have been detected in many solid tumors [
73]. Notably, a low expression of NPRL2, one of the GATOR1 components, in different types of lung cancers is correlated with cisplatin resistance [
74,
75]. The overexpression of NPRL2 in NPRL2-deficient and cisplatin-resistant NSCLC cells reactivates cellular response to cisplatin and promotes tumor suppression activity in vitro and in mouse models [
74]. The reason for this resistance is still not clear, but it could be related to the role of NPRL2 in DNA damage response [
75,
76].
4. Autophagy in Cisplatin Resistance
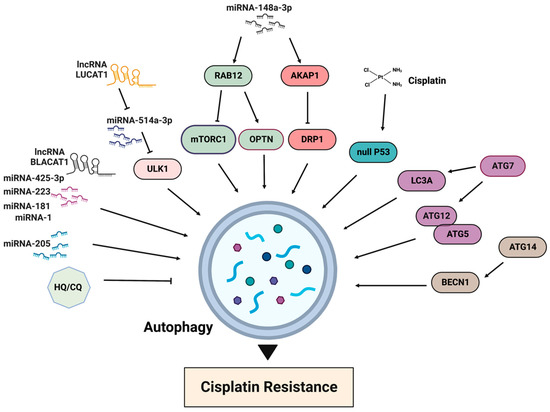
The activation of the mTORC1 pathway results in autophagy inhibition, while cisplatin treatment generally promotes autophagy (
Figure 6). One of the first reports about the association between autophagy and cisplatin resistance dates back to 2010, when it was demonstrated that the acquired cisplatin resistance in lung adenocarcinoma cells A549 was associated with elevated autophagy [
86]. The inhibition of autophagy is often observed in cisplatin-sensitive cells, whereas the basal level of autophagy is elevated in cisplatin-resistant cells. Accordingly, the suppression of autophagy, for example by chloroquine, increases drug toxicity and can improve sensitivity in cisplatin-resistant cancer cells [
87]. A recently published resource database of genes associated with platinum resistance in cancer demonstrates that genes involved in the production of autophagosomes, including ATG5, ATG7, ATG12, ATG14 and BECN1, promote platinum resistance [
88]. In the same line, an elevated expression of LC3A was shown to be associated with platinum resistance and a worse prognosis in ovarian clear cell carcinomas [
89]. Thus, the inhibition of autophagy can be considered as a strategy for improving cisplatin sensitivity.
Figure 6. Main factors of autophagy involved in cisplatin resistance. HQ/CQ: chloroquine and hydroxychloroquine.
Autophagy can also have a cytoprotective function, which is particularly important in the context of adverse effects during cisplatin treatment. Notably, up to 30% of patients receiving cisplatin develop acute kidney injury (AKI), leading to a rapid loss of renal function or renal failure. Autophagy is activated in renal tubules to protect against neurotoxicity during the acute phase. On the other hand, the sustained activation of autophagy will limit kidney repair. Thus, it is important to manipulate autophagy differently at the beginning and after the cisplatin treatment, in order to protect kidneys and allow for their effective recovery [
90].
Following cisplatin treatment, autophagy induction is detected in both cisplatin-sensitive and resistant cancer cells. Therefore, drug resistance is not necessarily a consequence of autophagy induction. It is important, though, to evaluate what type of autophagy is induced upon drug treatment because autophagy can participate in both cell survival (cytoprotective autophagy) and cell death (cytotoxic autophagy) [
92].
The inhibition of autophagy via different micro-RNAs also sensitized gastric cancer cells to cisplatin [
99,
100]. Thus, miR-148a-3p modulates cisplatin sensitivity by simultaneously regulating RAB12-mediated autophagy and AKAP1-mediated mitochondrial fission [
100]. RAB12 GTPase, a member of the RAS oncogene family, induces autophagy by inhibiting mTORC1 activity [
101] and accelerating autolysosome maturation [
102]. RAB12 also interacts with optineurin (OPTN), an important mitophagy receptor. The suppression of mTORC1 by RAB12 facilitates early autophagosome formation to protect gastric cancer cells from cisplatin-induced cell death. miR-148a-3p can significantly reduce autophagic flux and autophagosome formation by regulating RAB12. A-kinase anchoring protein 1 (AKAP1) is upregulated in cisplatin-resistant gastric cancer tissues and antagonizes cisplatin-induced mitochondrial fission by the phosphorylation of dynamin-related proteins 1 (DRP1), an important mitochondrial fission factor [
100]. Because AKAP1 restrains mitochondrial fission and reinforces cisplatin resistance in gastric cancer cells, targeting this protein by miR-148a-3 sensitizes cells to cisplatin. The role of DRP1 in cisplatin resistance is somewhat controversial and can be the opposite in different cancers. For example, in cisplatin-resistant ovarian cancer cell lines SKOV3, DRP1 expression was downregulated, and the knockdown of DRP1 in parental sensitive cell lines provoked cisplatin resistance [
11].
5. Conclusions
Cisplatin resistance is a significant challenge in cancer treatment, leading to decreased efficacy and poorer patient outcomes. Recent studies on the involvement of the mTORC1 pathway and autophagy in this resistance mechanism offer potential avenues for therapeutic intervention. Many mTORC1 signaling components and effectors are frequently deregulated or altered in different cancers. Cisplatin-resistant tumors often have a constitutive activation of mTORC1 signaling. Targeting the mTORC1 pathway with inhibitors has shown promising results in restoring cisplatin sensitivity, sensitizing resistant cells to the drug. Autophagy has emerged as another important player in cisplatin resistance because it can have a prosurvival role by protecting cancer cells from cisplatin-induced stress. Combination therapies that inhibit mTORC1 while modulating autophagy may provide a more effective strategy to sensitize resistant cancer cells to cisplatin.
This entry is adapted from the peer-reviewed paper 10.3390/ijms241310651