 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | John Mieyal | -- | 3046 | 2023-08-07 20:56:00 | | | |
| 2 | Lindsay Dong | Meta information modification | 3046 | 2023-08-08 03:18:08 | | |
Video Upload Options
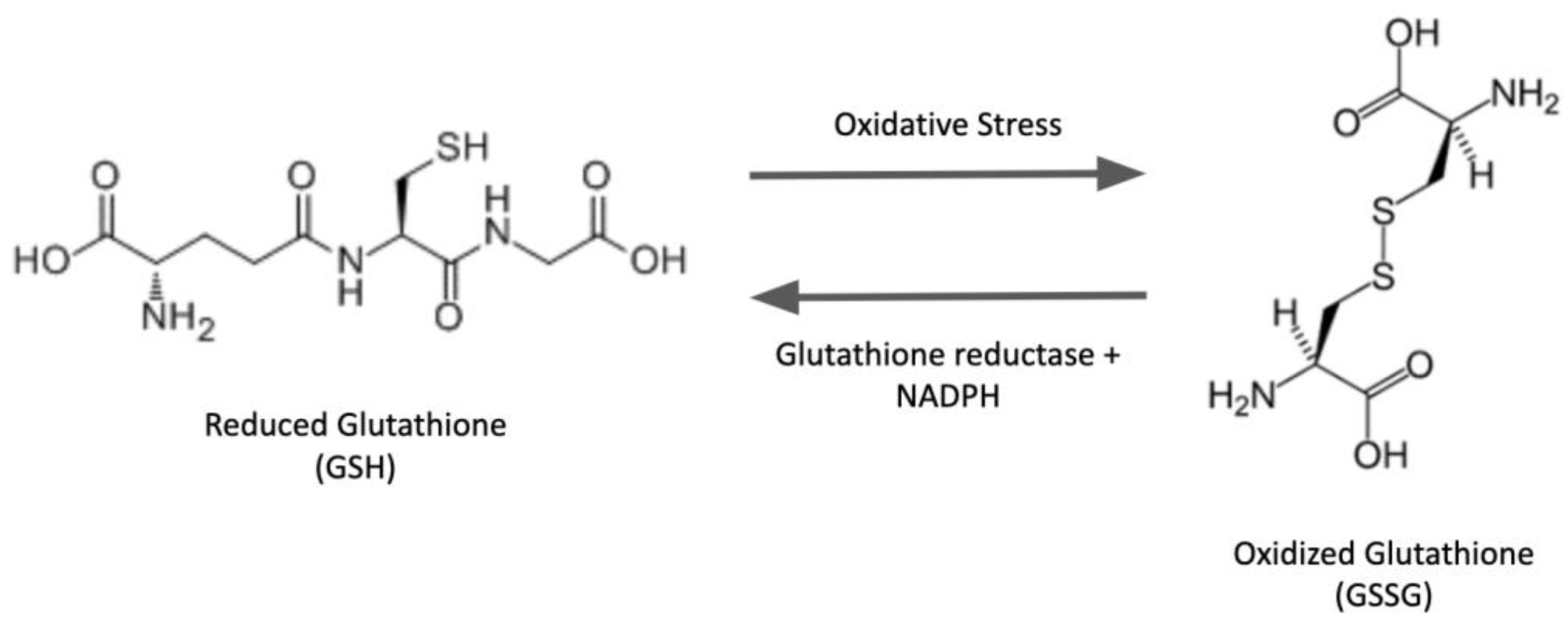
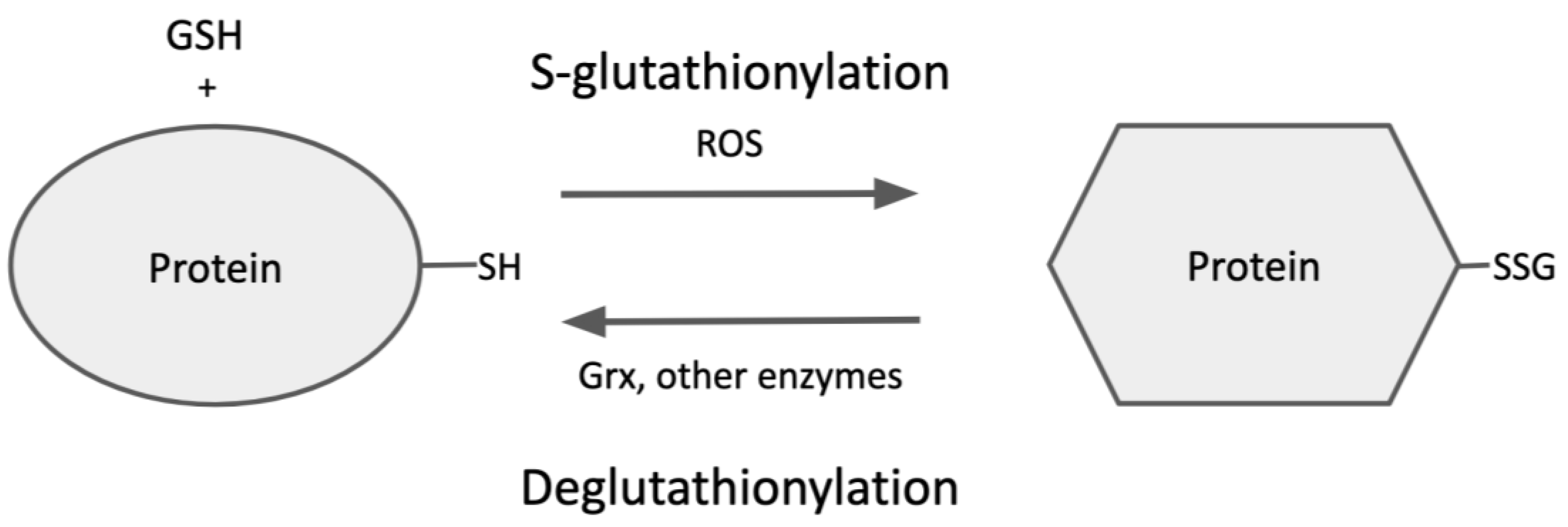
The tripeptide glutathione (GSH) is the most abundant non-enzymatic antioxidant/nucleophilic molecule in cells. In addition to various metabolic reactions involving GSH and its oxidized counterpart GSSG, oxidative post-translational modification (PTM) of proteins has been a focal point of keen interest in the redox field over the last few decades. In particular, the S-glutathionylation of proteins (protein-SSG formation), i.e., mixed disulfides between GSH and protein thiols, has been studied extensively. This reversible PTM can act as a regulatory switch to interconvert inactive and active forms of proteins, thereby mediating cell signaling and redox homeostasis. The unique architecture of the GSH molecule enhances its relative abundance in cells and contributes to the glutathionyl specificity of the primary catalytic activity of the glutaredoxin enzymes, which play central roles in redox homeostasis and signaling, and in iron metabolism in eukaryotes and prokaryotes under physiological and pathophysiological conditions. The class-1 glutaredoxins are characterized as cytosolic GSH-dependent oxidoreductases that catalyze reversible protein S-glutathionylation specifically, thereby contributing to the regulation of redox signal transduction and/or the protection of protein thiols from irreversible oxidation.
1. Overview
2. Glutaredoxin Enzymes
3. Protein S-Glutathionylation
4. Deglutathionylation of Protein-SSG
5. Highlights of the Special Issue on Glutathione and Glutaredoxin
Two articles [41][42] feature the specific roles of Grx1 in liver fibrosis and lung fibrosis. Importantly, the data presented in these papers suggest a potential therapeutic role for Grx1 as an anti-fibrotic agent. Thus, Reiko Matsui and her coworkers [41] showed that the overexpression of Grx1 inhibits age-induced hepatic apoptosis and liver fibrosis in mice. On the other hand, high-fat and high-fructose diet-induced non-alcoholic steatohepatitis (NASH) leads to the downregulation of Grx1 and higher levels of S-glutathionylated proteins in the liver; overexpression of Grx-1 significantly decreases the expression of Zbtb16 and leads to the reversal of NASH progression by attenuating inflammatory and fibrotic processes. Although the primary role of Zbtb16 in hepatocytes is unknown, the current study highlights it as an important redox-sensitive protein, whose expression is regulated by Grx1. Certainly, further study of Zbtb16 function is warranted.
Yvonne Janssen-Heininger and her coworkers [42] reported that Grx1 activity was directly correlated with lung function, whereas protein-SSG accumulation was inversely correlated with lung function in subjects with idiopathic pulmonary fibrosis. Epithelial cells lacking Grx1 were more susceptible to Fas-ligand-induced apoptosis and displayed elevated FAS-SSG compared to wild-type controls, whereas the overexpression of Grx1 attenuated epithelial cell apoptosis in association with diminished Fas-SSG. Several metabolites in the purine, creatine, and other metabolic pathways, including inosine monophosphate, spermidine, and others, were consistently released from multiple cell types subjected to various apoptotic stimuli, including Fas. These findings establish a link between Grx1 activity and the modulation of multiple pathways that regulate the synthesis and utilization of diverse metabolites released by apoptotic cells.
David Davis, Robert Yarchoan, and their coworkers [43] reported that protein S-glutathionylation regulates retroviral protease activity, including human immunodeficiency virus type 1 (HIV-1), human T-cell leukemia virus (HTLV-1), and SARS-CoV-2 proteases. In general, particular proteases of each virus are required for viral maturation, and the protease activities are dependent on the dimeric forms of the enzymes, which can be altered by site-selective S-glutathionylation. For example, HIV-1 protease contains two cysteine residues, Cys67 and Cys95, with low pKa values. S-glutathionylation of Cys67 (C95A-mutant protease) increased the activity by two-fold. On the contrary, S-glutathionylation of Cys95 completely inhibits the activity by disrupting the dimerization of the protease. The oxidation of Cys95 in immature virions impaired viral maturation, and this effect can be reversed by disulfide reduction. Grx1 catalyzes the deglutathionylation of Cys95 and restores protease activity much more efficiently than it deglutathionylates Cys67. Likewise, HTLV-1 protease activity can be regulated by S-glutathionylation and activity restored by Grx1. The S-glutathionylation of Cys95 in HIV-1 protease and Cys109 in HLTV-1 protease sterically interfere with beta-sheet formation and dimerization, according to crystal structure studies.
In analogous studies of Alzheimer’s disease (AD), increased S-glutathionylation of proteins has been observed in brain samples from AD patients, and actin-SSG is one of the target proteins. Importantly, the regulation of the dynamic polymerization of actin is vital to the function of neural synapses, affecting memory and learning, and it is noteworthy that overexpression of Grx1 in primary cortical culture leads to the restoration of F-actin nano-assembly and spine morphology.
References
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153.
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12.
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502.
- Pirie, N.W.; Pinhey, K.G. The Titration Curve of Glutathione. J. Biol. Chem. 1929, 184, 321–333.
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988.
- Outten, C.E.; Albetel, A.N. Iron sensing and regulation in Saccharomyces cerevisiae: Ironing out the mechanistic details. Curr. Opin. Microbiol. 2013, 16, 662–668.
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266.
- Couturier, J.; Przybyla-Toscano, J.; Roret, T.; Didierjean, C.; Rouhier, N. The roles of glutaredoxins ligating Fe-S clusters: Sensing, transfer or repair functions? Biochim. Biophys. Acta 2015, 1853, 1513–1527.
- Berndt, C.; Lillig, C.H. Glutathione, Glutaredoxins, and Iron. Antioxid. Redox Signal. 2017, 27, 1235–1251.
- Talib, E.A.; Outten, C.E. Iron-sulfur cluster biogenesis, trafficking, and signaling: Roles for CGFS glutaredoxins and BolA proteins. Biochimica et biophysica acta. Mol. Cell Res. 2021, 1868, 118847.
- Berndt, C.; Christ, L.; Rouhier, N.; Mühlenhoff, U. Glutaredoxins with iron-sulphur clusters in eukaryotes—Structure, function and impact on disease. Biochimica et biophysica acta. Bioenergetics 2021, 1862, 148317.
- Liedgens, L.; Zimmermann, J.; Wäschenbach, L.; Geissel, F.; Laporte, H.; Gohlke, H.; Morgan, B.; Deponte, M. Quantitative assessment of the determinant structural differences between redox-active and inactive glutaredoxins. Nat. Commun. 2020, 11, 1725.
- Yang, Y.; Jao, S.C.; Nanduri, S.; Starke, D.W.; Mieyal, J.J.; Qin, J. Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity. Biochemistry 1998, 37, 17145–17156.
- Saaranen, M.J.; Salo, K.E.; Latva-Ranta, M.K.; Kinnula, V.L.; Ruddock, L.W. The C-terminal active site cysteine of Escherichia coli glutaredoxin 1 determines the glutathione specificity of the second step of peptide deglutathionylation. Antioxid. Redox Signal. 2009, 11, 1819–1828.
- Trnka, D.; Engelke, A.D.; Gellert, M.; Moseler, A.; Hossain, M.F.; Lindenberg, T.T.; Pedroletti, L.; Odermatt, B.; de Souza, J.V.; Bronowska, A.K.; et al. Molecular basis for the distinct functions of redox-active and FeS-transfering glutaredoxins. Nat. Commun. 2020, 11, 3445.
- Couturier, J.; Jacquot, J.P.; Rouhier, N. Evolution and diversity of glutaredoxins in photosynthetic organisms. Cell. Mol. Life Sci. CMLS 2009, 66, 2539–2557.
- Gutsche, N.; Thurow, C.; Zachgo, S.; Gatz, C. Plant-specific CC-type glutaredoxins: Functions in developmental processes and stress responses. Biol. Chem. 2015, 396, 495–509.
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15.
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619.
- Moran, L.K.; Gutteridge, J.M.; Quinlan, G.J. Thiols in cellular redox signalling and control. Curr. Med. Chem. 2001, 8, 763–772.
- Leonberg, A.K.; Chai, Y.C. The functional role of cysteine residues for c-Abl kinase activity. Mol. Cell. Biochem. 2007, 304, 207–212.
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391.
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157.
- Marino, S.M.; Gladyshev, V.N. Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 2012, 287, 4419–4425.
- Pace, N.J.; Weerapana, E. A competitive chemical-proteomic platform to identify zinc-binding cysteines. ACS Chem. Biol. 2014, 9, 258–265.
- Reddie, K.G.; Carroll, K.S. Expanding the functional diversity of proteins through cysteine oxidation. Curr. Opin. Chem. Biol. 2008, 12, 746–754.
- Go, Y.M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245.
- Requejo, R.; Hurd, T.R.; Costa, N.J.; Murphy, M.P. Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 2010, 277, 1465–1480.
- Ferrer-Sueta, G.; Manta, B.; Botti, H.; Radi, R.; Trujillo, M.; Denicola, A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem. Res. Toxicol. 2011, 24, 434–450.
- Dalle-Donne, I.; Milzani, A.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 445–473.
- Gupta, V.; Carroll, K.S. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta 2014, 1840, 847–875.
- Harmel, R.; Fiedler, D. Features and regulation of non-enzymatic post-translational modifications. Nat. Chem. Biol. 2018, 14, 244–252.
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700.
- Zimmermann, J.; Oestreicher, J.; Hess, S.; Herrmann, J.M.; Deponte, M.; Morgan, B. One cysteine is enough: A monothiol Grx can functionally replace all cytosolic Trx and dithiol Grx. Redox Biol. 2020, 36, 101598.
- Chrestensen, C.A.; Starke, D.W.; Mieyal, J.J. Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J. Biol. Chem. 2000, 275, 26556–26565.
- Jung, C.H.; Thomas, J.A. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Arch. Biochem. Biophys. 1996, 335, 61–72.
- Gravina, S.A.; Mieyal, J.J. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry 1993, 32, 3368–3376.
- Gallogly, M.M.; Starke, D.W.; Leonberg, A.K.; Ospina, S.M.; Mieyal, J.J. Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: Implications for intracellular roles. Biochemistry 2008, 47, 11144–11157.
- Gallogly, M.M.; Starke, D.W.; Mieyal, J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox Signal. 2009, 11, 1059–1081.
- Lillig, C.H.; Berndt, C.; Vergnolle, O.; Lönn, M.E.; Hudemann, C.; Bill, E.; Holmgren, A. Characterization of human glutaredoxin 2 as iron-sulfur protein: A possible role as redox sensor. Proc. Natl. Acad. Sci. USA 2005, 102, 8168–8173.
- Tsukahara, Y.; Ferran, B.; Minetti, E.T.; Chong, B.S.H.; Gower, A.C.; Bachschmid, M.M.; Matsui, R. Administration of Glutaredoxin-1 Attenuates Liver Fibrosis Caused by Aging and Non-Alcoholic Steatohepatitis. Antioxidants 2022, 11, 867.
- Corteselli, E.; Aboushousha, R.; Janssen-Heininger, Y. S-Glutathionylation-Controlled Apoptosis of Lung Epithelial Cells; Potential Implications for Lung Fibrosis. Antioxidants 2022, 11, 1789.
- Davis, D.A.; Bulut, H.; Shrestha, P.; Mitsuya, H.; Yarchoan, R. Regulation of Retroviral and SARS-CoV-2 Protease Dimerization and Activity through Reversible Oxidation. Antioxidants 2022, 11, 2054.
- Wang, K.; Hirschenson, J.; Moore, A.; Mailloux, R.J. Conditions Conducive to the Glutathionylation of Complex I Subunit NDUFS1 Augment ROS Production following the Oxidation of Ubiquinone Linked Substrates, Glycerol-3-Phosphate and Proline. Antioxidants 2022, 11, 2043.
- Saaranen, M.J.; Alanen, H.I.; Salo, K.E.H.; Nji, E.; Kärkkäinen, P.; Schmotz, C.; Ruddock, L.W. Introduction of a More Glutaredoxin-like Active Site to PDI Results in Competition between Protein Substrate and Glutathione Binding. Antioxidants 2022, 11, 1920.
- Li, X.; Zhang, T.; Day, N.J.; Feng, S.; Gaffrey, M.J.; Qian, W.J. Defining the S-Glutathionylation Proteome by Biochemical and Mass Spectrometric Approaches. Antioxidants 2022, 11, 2272.
- Chakraborty, S.; Sircar, E.; Bhattacharyya, C.; Choudhuri, A.; Mishra, A.; Dutta, S.; Bhatta, S.; Sachin, K.; Sengupta, R. S-Denitrosylation: A Crosstalk between Glutathione and Redoxin Systems. Antioxidants 2022, 11, 1921.

