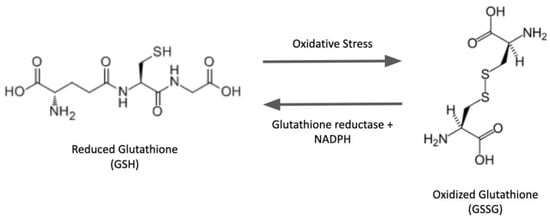
The tripeptide glutathione (GSH) is the most abundant non-enzymatic antioxidant/nucleophilic molecule in cells. In addition to various metabolic reactions involving GSH and its oxidized counterpart GSSG, oxidative post-translational modification (PTM) of proteins has been a focal point of keen interest in the redox field over the last few decades. In particular, the S-glutathionylation of proteins (protein-SSG formation), i.e., mixed disulfides between GSH and protein thiols, has been studied extensively. This reversible PTM can act as a regulatory switch to interconvert inactive and active forms of proteins, thereby mediating cell signaling and redox homeostasis. The unique architecture of the GSH molecule enhances its relative abundance in cells and contributes to the glutathionyl specificity of the primary catalytic activity of the glutaredoxin enzymes, which play central roles in redox homeostasis and signaling, and in iron metabolism in eukaryotes and prokaryotes under physiological and pathophysiological conditions. The class-1 glutaredoxins are characterized as cytosolic GSH-dependent oxidoreductases that catalyze reversible protein S-glutathionylation specifically, thereby contributing to the regulation of redox signal transduction and/or the protection of protein thiols from irreversible oxidation.
- glutathione
- glutaredoxin
- glutathionylation
- redox homeostasis
- redox signaling
- oxidative stress
1. Overview
2. Glutaredoxin Enzymes
The glutaredoxin oxidoreductase enzymes play central roles in redox homeostasis and signaling, and iron metabolism in eukaryotes and prokaryotes, under physiological and pathophysiological conditions [5][6][7][8][9][10][11][12][7,8,9,10,11,12,13,14]. The glutaredoxins are members of the thioredoxin superfamily and are structurally quite similar to thioredoxins and the N-terminal domain of glutathione transferases [7][9]; however, glutaredoxins have distinct catalytic properties. Most glutaredoxins belong to two major classes. Class I (Grx) includes the glutathione-dependent thiol:disulfide oxidoreductases [5][7][12][13][14][7,9,14,15,16], and class II includes the Grx-like proteins (Glp), which are inactive in standard oxidoreductase assays but serve as iron sensors, playing a critical role in glutathione-dependent delivery of iron–sulfur (Fe-S) clusters [6][8][9][10][15][8,10,11,12,17]. There are other minor Grx subfamilies, including several plant-specific Grx isoforms [16][18], which exert additional functions, such as the transcriptional regulation of petal development in flowers [17][19].3. Protein S-Glutathionylation
Although historically viewed as harmful byproducts of metabolism that are scavenged by antioxidants, reactive oxygen species (ROS) are also known to act as intracellular second messengers in redox-signaling pathways [18][19][20,21]. ROS serve this redox regulation function by modifying specific signaling proteins. In particular, ROS-mediated reversible oxidative modifications of the thiol moieties of protein-cysteine residues are characterized by their ability to modulate the protein activities, thereby propagating signal transduction and biological responses [20][21][22,23]. Such post-translational modifications of reactive protein-thiols include sulfenic acid formation (protein-SOH), nitrosylation (protein-SNO), S-glutathionylation (protein-SSG), and others. Considering the relative abundance of GSH in cells and the relative reactivity of various modified cysteine intermediates, it is likely that protein-SSG may represent the preponderant form of protein–cysteine modification [22][24]. Although cysteine is one of the least abundant amino acids, it stands out as functionally distinct [23][25]. Even though it has a rather low occurrence in proteins in general, cysteine is often found in the functional sites of proteins, where it plays key roles in catalysis, regulation, secondary structure, etc. [24][26]. Moreover, cysteine residues often constitute metal-binding sites on proteins, facilitating the action of metal ions as cofactors [25][27]. These characteristic properties of cysteine residues are dependent on the physical nature and chemistry of the sulfhydryl moiety. Thus, the nucleophilicity of the thiol group is responsible for cysteine’s role in catalysis by enzymes like kinases and phosphatases. The redox reactivity of the thiol group enables cysteine to participate in structural thiol-disulfide interchange reactions affecting protein stability, and in oxidative posttranslational modifications that regulate function and propagate signaling pathways [26][28]. About 214,000 cysteines are encoded in the human genome [27][31]. Proteins may have exposed cysteine residues on the surface within the aqueous environment [28][32], or may be embedded deep within the more hydrophobic globular domains. In an aqueous milieu, the thiol group of cysteine is prone to deprotonation, in equilibrium with the negatively charged thiolate moiety. Both the protonated and unprotonated forms of cysteine have non-bonded pairs of electrons, consistent with nucleophilicity, but the thiolate anions are much more reactive. The ratio (thiol/thiolate) at any pH condition is related to the thiol pKa, which for a typical cysteine residue is near 8.5 [23][25]; accordingly, only a small fraction of cysteine residues would be negatively charged at physiological pH (7.4). However, the local microenvironment (e.g., neighboring cations) can strongly affect the pKa values of protein thiols over a wide range [24][26]. Although a lower pKa value corresponds to a higher thiolate amount at neutral pH, it is important to note that the relative rates of cysteine-mediated reactions also depend on other factors [29][33]. Among the mechanisms of formation of protein-SSG adducts, thiol-disulfide exchange is one of the most extensively studied [5][7]; however, it relies primarily on the redox state of cellular glutathione. According to this mechanism, the intracellular GSH/GSSG ratio dictates the extent of protein S-glutathionylation ([Protein-SSG]/[Protein-SH]), and the equilibrium constant for the reaction (Kmix, Equation (1)) corresponds to the oxidation potential for the formation of the mixed disulfide (protein-SSG): Protein-SH + GSSG ⇌ Protein-SSG + GSH, The Kmix value for most cysteine residues is approximately 1.0, so the GSH/GSSG ratio would have to be decreased greatly in order to favor the formation of protein-SSG [5][7]. However, as described above, the cytosolic GSH/GSSG ratio usually remains very high, even under pronounced oxidative stress conditions [30][34], rendering the thiol-disulfide exchange mechanism with GSSG as the mediator thermodynamically unfavorable. Another important mechanism that may mediate protein-SSG formation in vivo involves another type of reactive thiol derivate, namely sulfenic acid. Several different oxidants, including hydrogen peroxide, alkyl hydroperoxides, peroxynitrite, hypochlorous acid, and chloramines, are implicated in mediating the conversion of protein-thiolates to sulfenic acids (protein-SOH) [31][41], but increased exposure to these oxidants can lead to further oxidation and irreversible modification (sulfinic and sulfonic acids), or promote reactions with neighboring thiols to form disulfides [31][41]. Sulfenic acid formation was found to be a regulatory mechanism for many proteins [23][25].4. Deglutathionylation of Protein-SSG
As mentioned above, S-glutathionylation of proteins is dynamic and reversible, so the steady-state level of protein-SSG under various conditions depends on the relative rates of glutathionylation (formation) and deglutathionylation (breakdown) (Figure 23), providing another level of regulation for cellular processes [32][53]. Specific binding sites for glutathione have been characterized on glutaredoxin, which is understood to be the primary catalyst of deglutathionylation [33][54].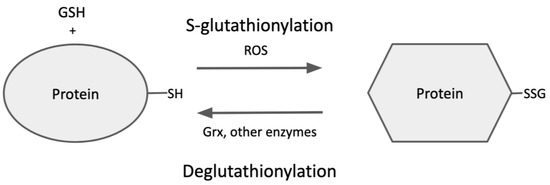
5. Highlights of the Special Issue on Glutathione and Glutaredoxin
Two articles [41][42][73,74] feature the specific roles of Grx1 in liver fibrosis and lung fibrosis. Importantly, the data presented in these papers suggest a potential therapeutic role for Grx1 as an anti-fibrotic agent. Thus, Reiko Matsui and her coworkers [41][73] showed that the overexpression of Grx1 inhibits age-induced hepatic apoptosis and liver fibrosis in mice. On the other hand, high-fat and high-fructose diet-induced non-alcoholic steatohepatitis (NASH) leads to the downregulation of Grx1 and higher levels of S-glutathionylated proteins in the liver; overexpression of Grx-1 significantly decreases the expression of Zbtb16 and leads to the reversal of NASH progression by attenuating inflammatory and fibrotic processes. Although the primary role of Zbtb16 in hepatocytes is unknown, the current study highlights it as an important redox-sensitive protein, whose expression is regulated by Grx1. Certainly, further study of Zbtb16 function is warranted.
Yvonne Janssen-Heininger and her coworkers [42][74] reported that Grx1 activity was directly correlated with lung function, whereas protein-SSG accumulation was inversely correlated with lung function in subjects with idiopathic pulmonary fibrosis. Epithelial cells lacking Grx1 were more susceptible to Fas-ligand-induced apoptosis and displayed elevated FAS-SSG compared to wild-type controls, whereas the overexpression of Grx1 attenuated epithelial cell apoptosis in association with diminished Fas-SSG. Several metabolites in the purine, creatine, and other metabolic pathways, including inosine monophosphate, spermidine, and others, were consistently released from multiple cell types subjected to various apoptotic stimuli, including Fas. These findings establish a link between Grx1 activity and the modulation of multiple pathways that regulate the synthesis and utilization of diverse metabolites released by apoptotic cells.
David Davis, Robert Yarchoan, and their coworkers [43][76] reported that protein S-glutathionylation regulates retroviral protease activity, including human immunodeficiency virus type 1 (HIV-1), human T-cell leukemia virus (HTLV-1), and SARS-CoV-2 proteases. In general, particular proteases of each virus are required for viral maturation, and the protease activities are dependent on the dimeric forms of the enzymes, which can be altered by site-selective S-glutathionylation. For example, HIV-1 protease contains two cysteine residues, Cys67 and Cys95, with low pKa values. S-glutathionylation of Cys67 (C95A-mutant protease) increased the activity by two-fold. On the contrary, S-glutathionylation of Cys95 completely inhibits the activity by disrupting the dimerization of the protease. The oxidation of Cys95 in immature virions impaired viral maturation, and this effect can be reversed by disulfide reduction. Grx1 catalyzes the deglutathionylation of Cys95 and restores protease activity much more efficiently than it deglutathionylates Cys67. Likewise, HTLV-1 protease activity can be regulated by S-glutathionylation and activity restored by Grx1. The S-glutathionylation of Cys95 in HIV-1 protease and Cys109 in HLTV-1 protease sterically interfere with beta-sheet formation and dimerization, according to crystal structure studies.
In analogous studies of Alzheimer’s disease (AD), increased S-glutathionylation of proteins has been observed in brain samples from AD patients, and actin-SSG is one of the target proteins. Importantly, the regulation of the dynamic polymerization of actin is vital to the function of neural synapses, affecting memory and learning, and it is noteworthy that overexpression of Grx1 in primary cortical culture leads to the restoration of F-actin nano-assembly and spine morphology.