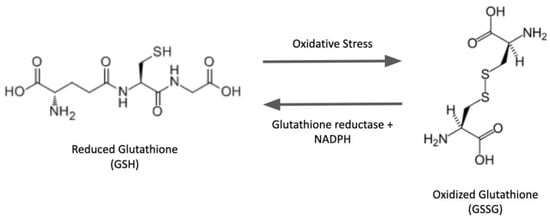
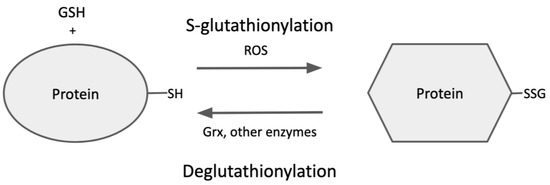
Glutaredoxin isoforms exist in various subcellular compartments in eukaryotes and also in prokaryotes. As described under
Section 1, mammalian glutaredoxins are broadly classified into two subfamilies based on their active site sequences and relative activities/functions, with Grx1 and Grx2 representing the major forms of each family [
56]. Human Grx1 is localized in the cytosol and mitochondrial intermembrane space, whereas Grx2 is primarily localized in mitochondria. Grx1 is better characterized and reported to catalyze most of the deglutathionylating activity in mammalian cells [
57,
58]. Although Grx1 is not an essential protein since knockout mice are viable with a life span similar to wild-type mice [
54], it is implicated broadly in cellular functions and defense against disease. For example, the level of protein S-glutathionylation is linked to the development of diseases in Grx1-knockout models [
54], and the high concentrations of protein-SSG could be reversed by the exogenous administration of recombinant Grx1.
Grx1 and Grx2 have both been shown to selectivity catalyze dethiolation of S-glutathionylated substrates with GSH as co-substrate. The glutaredoxin substrate specificity was determined by a carefully designed experiment using various protein mixed-disulfide substrates with glutathione- and non-glutathione-containing thiols [
69]. Grx effectively catalyzed only the glutathione-containing substrates but was ineffective for other substrates. These two isozymes share an analogous catalytic mechanism for deglutathionylation involving a nucleophilic, double-displacement (ping-pong) sequence, wherein only the N-terminal cysteine residue of the active-site CPYC motif participates in catalysis (monothiol mechanism) [
70]. The main distinction between them is the decreased catalytic efficiency (k
cat/K
M) of Grx2, primarily due to a decreased k
cat [
70]. The lower Kcat of Grx2 is because of the catalytic cysteine’s higher pKa and a decreased nucleophilicity enhancement of the second substrate, GSH [
70]. Like Grx1, Grx2 exhibits GS-thiyl radical (GS
•) scavenging activity, promoting the S-glutathionylation of various proteins [
70].
Human glutaredoxin 2 (hGrx2) was the first member of the glutaredoxin family identified as an iron–sulfur protein. The Fe-S cofactor was shown to be bridged between two monomers via the N-terminal active site cysteine residues and two non-covalently bound GSH molecules [
68]. The bound GSH was in exchange with the free GSH pool and played an essential role in stabilizing the Fe-S cluster. The dimeric form of hGrx2 was enzymatically inactive [
68]. Under oxidative stress, an altered GSH/GSSG ratio limiting reduced GSH availability to maintain the Fe-S coordination resulted in the degradation of the cluster and formation of the enzymatically active Grx2 monomer. These results were interpreted to suggest that the Fe-S cluster functions as a redox sensor for Grx2 activity under oxidative stress. As explained earlier, other members of the broad glutaredoxin family have been characterized as participating in iron–sulfur homeostasis [
10,
11,
17].
5. Highlights of the Special Issue on Glutathione and Glutaredoxin
Two articles [73,74] feature the specific roles of Grx1 in liver fibrosis and lung fibrosis. Importantly, the data presented in these papers suggest a potential therapeutic role for Grx1 as an anti-fibrotic agent. Thus, Reiko Matsui and her coworkers [73] showed that the overexpression of Grx1 inhibits age-induced hepatic apoptosis and liver fibrosis in mice. On the other hand, high-fat and high-fructose diet-induced non-alcoholic steatohepatitis (NASH) leads to the downregulation of Grx1 and higher levels of S-glutathionylated proteins in the liver; overexpression of Grx-1 significantly decreases the expression of Zbtb16 and leads to the reversal of NASH progression by attenuating inflammatory and fibrotic processes. Although the primary role of Zbtb16 in hepatocytes is unknown, the current study highlights it as an important redox-sensitive protein, whose expression is regulated by Grx1. Certainly, further study of Zbtb16 function is warranted.
Yvonne Janssen-Heininger and her coworkers [74] reported that Grx1 activity was directly correlated with lung function, whereas protein-SSG accumulation was inversely correlated with lung function in subjects with idiopathic pulmonary fibrosis. Epithelial cells lacking Grx1 were more susceptible to Fas-ligand-induced apoptosis and displayed elevated FAS-SSG compared to wild-type controls, whereas the overexpression of Grx1 attenuated epithelial cell apoptosis in association with diminished Fas-SSG. Several metabolites in the purine, creatine, and other metabolic pathways, including inosine monophosphate, spermidine, and others, were consistently released from multiple cell types subjected to various apoptotic stimuli, including Fas. These findings establish a link between Grx1 activity and the modulation of multiple pathways that regulate the synthesis and utilization of diverse metabolites released by apoptotic cells.
David Davis, Robert Yarchoan, and their coworkers [76] reported that protein S-glutathionylation regulates retroviral protease activity, including human immunodeficiency virus type 1 (HIV-1), human T-cell leukemia virus (HTLV-1), and SARS-CoV-2 proteases. In general, particular proteases of each virus are required for viral maturation, and the protease activities are dependent on the dimeric forms of the enzymes, which can be altered by site-selective S-glutathionylation. For example, HIV-1 protease contains two cysteine residues, Cys67 and Cys95, with low pKa values. S-glutathionylation of Cys67 (C95A-mutant protease) increased the activity by two-fold. On the contrary, S-glutathionylation of Cys95 completely inhibits the activity by disrupting the dimerization of the protease. The oxidation of Cys95 in immature virions impaired viral maturation, and this effect can be reversed by disulfide reduction. Grx1 catalyzes the deglutathionylation of Cys95 and restores protease activity much more efficiently than it deglutathionylates Cys67. Likewise, HTLV-1 protease activity can be regulated by S-glutathionylation and activity restored by Grx1. The S-glutathionylation of Cys95 in HIV-1 protease and Cys109 in HLTV-1 protease sterically interfere with beta-sheet formation and dimerization, according to crystal structure studies.
In analogous studies of Alzheimer’s disease (AD), increased S-glutathionylation of proteins has been observed in brain samples from AD patients, and actin-SSG is one of the target proteins. Importantly, the regulation of the dynamic polymerization of actin is vital to the function of neural synapses, affecting memory and learning, and it is noteworthy that overexpression of Grx1 in primary cortical culture leads to the restoration of F-actin nano-assembly and spine morphology.
Continuing the focus on mitochondria as an engine of oxidative stress, Ryan Mailloux and coworkers in an original research contribution to the Special Issue [
79] reported S-glutathionylation of the NDUFS1 subunit of Complex 1 of the electron transport chain in liver mitochondria. This modification caused inhibition of Complex I activity and increased the generation of hydrogen peroxide, using glycerol-3-phosphate or proline as fuel during reverse electron transfer. Adding a reducing agent to the reaction system was found to reverse both the inhibition and the ROS accumulation. Interestingly, two other mitochondria proteins, glycerol-3-phosphate dehydrogenase and proline dehydrogenase, were not S-glutathionylated under conditions similar to those that led to NDUFS1-SSG formation, even though these two proteins also produce ROS. Complex III also undergoes S-glutathionylation with an effect similar to that of Complex I, but whether this effect is due to the Complex III modification or glutathionylation of complexes upstream from ubquinone–cytochrome C oxidoreductase remains to be elucidated.
Both Grx and protein disulfide isomerase (PDI) belong to the thioredoxin superfamily, contain CXXC sequences at their respective active sites, and utilize glutathione as a co-substrate. Although these two proteins have a similar 3D structure and thiol-disulfide catalytic mechanism, their cellular environments are quite different; thus, Grx catalyzes the reduction of glutathionyl mixed disulfides, whereas PDI catalyzes the formation of intramolecular disulfides. Ruddock and coworkers have been studying these two enzymes, and they contributed an original research article to this compendium [
80] in which they reported the mutation of the PDI CXXC motif, converting histidine to tyrosine or phenylalanine, thereby changing the signature sequence to be more similar to that of class I glutaredoxins. These substitutions for histidine were found to change the binding affinity of PDI for its protein substrates and glutathione.
A very important aspect of understanding the impact of post-translational modification of protein–cysteine residues on the functional consequences is the validity and accuracy of the analytical methods used to identify the PTMs. In this compendium, Wei-Jun Qian and coworkers [
81] have contributed a review article focused on the current biochemical and analytical approaches to characterize protein S-glutathionylation at both the proteome level and with individual proteins, including a perspective on studies of the functional impacts. Importantly, they highlight the challenges of some of the methods and consider ways to overcome these difficulties in future work. For example, it is difficult to directly verify the S-glutathionylation of specific cysteinyl moieties in their native environment in situ using mass spectroscopic (MS) approaches. The glutathionyl moiety on a Cys residue can undergo fragmentation in the MS/MS mode of analysis, leading to neutral losses via cleavage of the peptide bond. Another challenge is interpreting the S-glutathionylation data due to the complexity of the oxidative modification of cysteine.
S-nitrosylation is a form of cysteine modification that has been broadly implicated in signal transduction, cellular homeostasis, and disease. Rajib Sengupta and coworkers have a special interest in S-nitrosylation as a regulatory mechanism. In their review that contributed to this Special Issue [
82], they focused on the mechanisms of denitrosylation of protein-SNO and the enzymes that catalyze it, analogous to the catalysis of deglutathionylation by glutaredoxin. S-nitrosylated proteins can be denitrosylated by GSH within the range of the physiological concentrations of GSH (5–10 mM), except for a few proteins, such as caspase 3. It is conceivable that those proteins that are resistant to dentrosylation by GSH alone may be more likely to be regulated by reversible S-nitrosylation, requiring enzymatic reversal. GSH denitrosylates proteins in two ways: by displacement of the NO moiety, forming protein-SSG, or by transnitrosylation, forming GS-NO. A key element in this mechanism is GS-NO reductase (GSNOR), which is a significant regulator of the relative cellular levels of protein-SNO and GS-NO. Thus, GSNOR does not directly denitrosylate proteins; instead, it decreases protein-SNO levels by depleting GS-NO. Caspase3-SNO, which is not denitrosylated by GSH/GSNOR, is reduced by the thioredoxin (Trx) system.