 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ali Mussa | -- | 5102 | 2023-08-07 10:54:17 | | | |
| 2 | Peter Tang | Meta information modification | 5102 | 2023-08-07 11:24:28 | | |
Video Upload Options
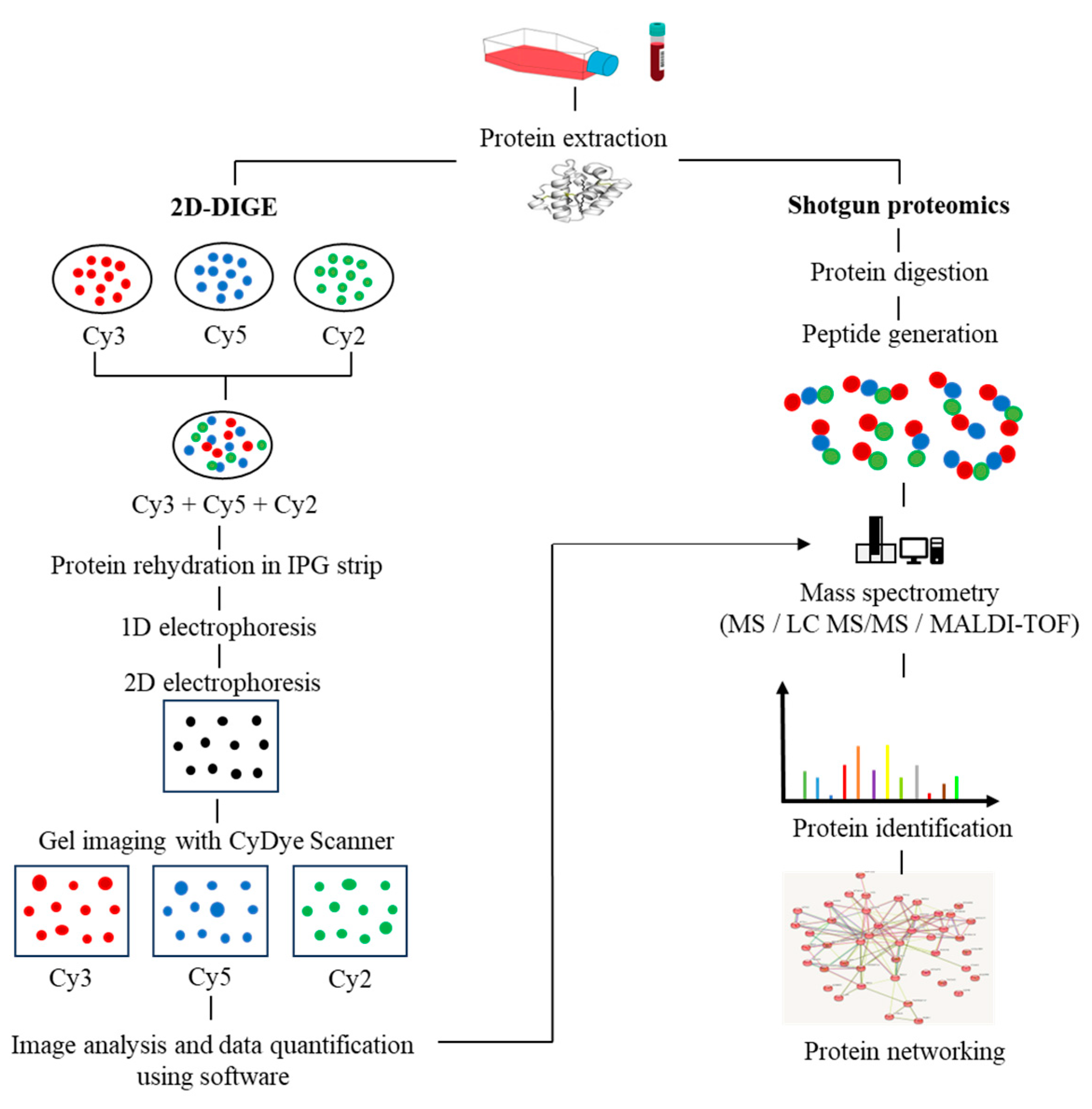
Multiple myeloma (MM) is an incurable hematologic malignancy. Most MM patients are diagnosed at a late stage because the early symptoms of the disease can be uncertain and nonspecific, often resembling other, more common conditions. Additionally, MM patients are commonly associated with rapid relapse and an inevitable refractory phase. MM is characterized by the abnormal proliferation of monoclonal plasma cells in the bone marrow. During the progression of MM, massive genomic alterations occur that target multiple signaling pathways and are accompanied by a multistep process involving differentiation, proliferation, and invasion. Moreover, the transformation of healthy plasma cell biology into genetically heterogeneous MM clones is driven by a variety of post-translational protein modifications (PTMs), which has complicated the discovery of effective treatments. PTMs have been identified as the most promising candidates for biomarker detection, and further research has been recommended to develop promising surrogate markers. Proteomics studies using mass spectrometry (MS) in conjunction with robust bioinformatics tools are an excellent way to learn more about protein changes and modifications during disease progression MM.
1. Introduction
2. Proteomics Breakthroughs in Protein Identification
3. Post-Translational Modifications in MM
4. Proteomics Alteration in MM
4.1. Monoclonal Gammopathy of Undetermined Significance (MGUS)
4.2. Smoldering Multiple Myeloma (SMM)
4.3. Multiple Myeloma (MM)
4.4. Myeloma Bone Disease
4.5. Relapse/Refractory MM
4.6. Extramedullary MM
5. Investigating Biomarkers in Drug-Resistant MM
6. Therapeutic Agents to Target Protein Signatures
References
- Hari, P.; Mateos, M.V.; Abonour, R.; Knop, S.; Bensinger, W.; Ludwig, H.; Song, K.; Hajek, R.; Moreau, P.; Siegel, D.S.; et al. Efficacy and safety of carfilzomib regimens in multiple myeloma patients relapsing after autologous stem cell transplant: ASPIRE and ENDEAVOR outcomes. Leukemia 2017, 31, 2630–2641.
- Tsang, M.; Le, M.; Ghazawi, F.M.; Cyr, J.; Alakel, A.; Rahme, E.; Lagacé, F.; Netchiporouk, E.; Moreau, L.; Zubarev, A.; et al. Multiple myeloma epidemiology and patient geographic distribution in Canada: A population study. Cancer 2019, 125, 2435–2444.
- Rajkumar, S.V. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 1086–1107.
- Prideaux, S.M.; Conway O’Brien, E.; Chevassut, T.J. The genetic architecture of multiple myeloma. Adv. Hematol. 2014, 2014, 864058.
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348.
- Manier, S.; Kawano, Y.; Bianchi, G.; Roccaro, A.M.; Ghobrial, I.M. Cell autonomous and microenvironmental regulation of tumor progression in precursor states of multiple myeloma. Curr. Opin. Hematol. 2016, 23, 426–433.
- Bezieau, S.; Devilder, M.C.; Avet-Loiseau, H.; Mellerin, M.P.; Puthier, D.; Pennarun, E.; Rapp, M.J.; Harousseau, J.L.; Moisan, J.P.; Bataille, R. High incidence of N and K-Ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis. Hum. Mutat. 2001, 18, 212–224.
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187.
- Ismail, N.H.; Mussa, A.; Zakaria, N.A.; Al-Khreisat, M.J.; Zahidin, M.A.; Ramli, N.N.; Mohammad, S.; Hassan, R.; Mohd Noor, N.H.; Iberahim, S.; et al. The Role of Epigenetics in the Development and Progression of Multiple Myeloma. Biomedicines 2022, 10, 2767.
- Zhan, F.; Hardin, J.; Kordsmeier, B.; Bumm, K.; Zheng, M.; Tian, E.; Sanderson, R.; Yang, Y.; Wilson, C.; Zangari, M.; et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002, 99, 1745–1757.
- Noone, A.-M.; Cronin, K.A.; Altekruse, S.F.; Howlader, N.; Lewis, D.R.; Petkov, V.I.; Penberthy, L. Cancer Incidence and Survival Trends by Subtype Using Data from the Surveillance Epidemiology and End Results Program, 1992–2013. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 632–641.
- Kumar, S.K.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Pandey, S.; Kapoor, P.; Dingli, D.; Hayman, S.R.; Leung, N.; et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia 2013, 28, 1122–1128.
- Mikhael, J.; Bhutani, M.; Cole, C.E. Multiple Myeloma for the Primary Care Provider: A Practical Review to Promote Earlier Diagnosis Among Diverse Populations. Am. J. Med. 2022, 136, 33–41.
- Nooka, A.K.; Kastritis, E.; Dimopoulos, M.A.; Lonial, S. Treatment options for relapsed and refractory multiple myeloma. Blood 2015, 125, 3085–3099.
- Cejalvo, M.J.; de la Rubia, J. Which therapies will move to the front line for multiple myeloma? Expert Rev. Hematol. 2017, 10, 383–392.
- Rajkumar, S.V. Multiple myeloma: Every year a new standard? Hematol. Oncol. 2019, 37 (Suppl. S1), 62–65.
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83.
- Woods, A.G.; Sokolowska, I.; Ngounou Wetie, A.G.; Channaveerappa, D.; Dupree, E.J.; Jayathirtha, M.; Aslebagh, R.; Wormwood, K.L.; Darie, C.C. Mass Spectrometry for Proteomics-Based Investigation. In Advancements of Mass Spectrometry in Biomedical Research; Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1140, pp. 1–26.
- Schirle, M.; Bantscheff, M.; Kuster, B. Mass Spectrometry-Based Proteomics in Preclinical Drug Discovery. Chem. Biol. 2012, 19, 72–84.
- Crutchfield, C.A.; Thomas, S.N.; Sokoll, L.J.; Chan, D.W. Advances in mass spectrometry-based clinical biomarker discovery. Clin. Proteom. 2016, 13, 1.
- Sperling, K. From proteomics to genomics. Electrophoresis 2001, 22, 2835–2837.
- Loo, J.A.; DeJohn, D.E.; Du, P.; Stevenson, T.I.; Ogorzalek Loo, R.R. Application of mass spectrometry for target identification and characterization. Med. Res. Rev. 1999, 19, 307–319.
- Yates, J.R., 3rd; McCormack, A.L.; Schieltz, D.; Carmack, E.; Link, A. Direct analysis of protein mixtures by tandem mass spectrometry. J. Protein Chem. 1997, 16, 495–497.
- Marouga, R.; David, S.; Hawkins, E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 2005, 382, 669–678.
- Barabási, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113.
- Zybailov, B.L.; Byrd, A.K.; Glazko, G.V.; Rahmatallah, Y.; Raney, K.D. Protein-protein interaction analysis for functional characterization of helicases. Methods 2016, 108, 56–64.
- Ge, F.; Xiao, C.-L.; Yin, X.-F.; Lu, C.-H.; Zeng, H.-L.; He, Q.-Y. Phosphoproteomic analysis of primary human multiple myeloma cells. J. Proteom. 2010, 73, 1381–1390.
- Ge, F.; Xiao, C.-L.; Bi, L.-J.; Tao, S.-C.; Xiong, S.; Yin, X.-F.; Li, L.-P.; Lu, C.-H.; Jia, H.-T.; He, Q.-Y. Quantitative phosphoproteomics of proteasome inhibition in multiple myeloma cells. PLoS ONE 2010, 5, e13095.
- Pollett, J.B.; Trudel, S.; Stern, D.; Li, Z.H.; Stewart, A.K. Overexpression of the myeloma-associated oncogene fibroblast growth factor receptor 3 confers dexamethasone resistance. Blood 2002, 100, 3819–3821.
- Bondt, A.; Rombouts, Y.; Selman, M.H.; Hensbergen, P.J.; Reiding, K.R.; Hazes, J.M.; Dolhain, R.J.; Wuhrer, M. Immunoglobulin G (IgG) Fab glycosylation analysis using a new mass spectrometric high-throughput profiling method reveals pregnancy-associated changes. Mol. Cell. Proteom. 2014, 13, 3029–3039.
- Van de Bovenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J. Immunol. 2016, 196, 1435–1441.
- Gornik, O.; Pavić, T.; Lauc, G. Alternative glycosylation modulates function of IgG and other proteins—Implications on evolution and disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 1318–1326.
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10, 104–115.
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919.
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880.
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell 2008, 29, 69–80.
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459.
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446.
- Ramachandran, S.; Chahwan, R.; Nepal, R.M.; Frieder, D.; Panier, S.; Roa, S.; Zaheen, A.; Durocher, D.; Scharff, M.D.; Martin, A. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc. Natl. Acad. Sci. USA 2009, 107, 809–814.
- Zhan, F.; Barlogie, B.; Arzoumanian, V.; Huang, Y.; Williams, D.R.; Hollmig, K.; Pineda-Roman, M.; Tricot, G.; van Rhee, F.; Zangari, M.; et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2006, 109, 1692–1700.
- Chng, W.J.; Kumar, S.; VanWier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.H.; Kim, S.; Mulligan, G.; Bryant, B.; et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res 2007, 67, 2982–2989.
- Jagani, Z.; Wiederschain, D.; Loo, A.; He, D.; Mosher, R.; Fordjour, P.; Monahan, J.; Morrissey, M.; Yao, Y.-M.; Lengauer, C.; et al. The Polycomb group protein Bmi-1 Is essential for the growth of multiple myeloma cells. Cancer Res 2010, 70, 5528–5538.
- Roy, P.; Sarkar, U.A.; Basak, S. The NF-κB activating pathways in multiple myeloma. Biomedicines 2018, 6, 59.
- Van Andel, H.; Kocemba, K.A.; de Haan-Kramer, A.; Mellink, C.H.; Piwowar, M.; Broijl, A.; Van Duin, M.; Sonneveld, P.; Maurice, M.; Kersten, M.J.; et al. Loss of CYLD expression unleashes Wnt signaling in multiple myeloma and is associated with aggressive disease. Oncogene 2016, 36, 2105–2115.
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell 2007, 12, 131–144.
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007, 12, 115–130.
- Zhao, Y.; Jensen, O.N. Modification-specific proteomics: Strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9, 4632–4641.
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618.
- Zhang, J.; Sprung, R.; Pei, J.; Tan, X.; Kim, S.; Zhu, H.; Liu, C.-F.; Grishin, N.V.; Zhao, Y. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteom. 2009, 8, 215–225.
- Ong, S.-E.; Mittler, G.; Mann, M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods 2004, 1, 119–126.
- Zhan, X.; Desiderio, D.M. Nitroproteins from a human pituitary adenoma tissue discovered with a nitrotyrosine affinity column and tandem mass spectrometry. Anal. Biochem. 2006, 354, 279–289.
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203.
- Pandey, A.; Podtelejnikov, A.V.; Blagoev, B.; Bustelo, X.R.; Mann, M.; Lodish, H.F. Analysis of receptor signaling pathways by mass spectrometry: Identification of Vav-2 as a substrate of the epidermal and platelet-derived growth factor receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 179–184.
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166.
- Van Kruijsbergen, I.; Mulder, M.P.C.; Uckelmann, M.; van Welsem, T.; de Widt, J.; Spanjaard, A.; Jacobs, H.; El Oualid, F.; Ovaa, H.; van Leeuwen, F. Strategy for Development of Site-Specific Ubiquitin Antibodies. Front. Chem. 2020, 8, 111.
- Vocadlo, D.J.; Hang, H.C.; Kim, E.J.; Hanover, J.A.; Bertozzi, C.R. A chemical approach for identifying O -GlcNAc-modified proteins in cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9116–9121.
- Baskin, J.M.; Prescher, J.A.; Laughlin, S.T.; Agard, N.J.; Chang, P.V.; Miller, I.A.; Lo, A.; Codelli, J.A.; Bertozzi, C.R. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797.
- Kho, Y.; Kim, S.C.; Jiang, C.; Barma, D.; Kwon, S.W.; Cheng, J.; Jaunbergs, J.; Weinbaum, C.; Tamanoi, F.; Falck, J.; et al. A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12479–12484.
- Sprung, R.; Nandi, A.; Chen, Y.; Kim, S.C.; Barma, D.; Falck, J.R.; Zhao, Y. Tagging-via-substrate strategy for probing O-GlcNAc modified proteins. J. Proteome Res. 2005, 4, 950–957.
- Kostiuk, M.A.; Corvi, M.M.; Keller, B.O.; Plummer, G.; Prescher, J.A.; Hangauer, M.J.; Bertozzi, C.R.; Rajaiah, G.; Falck, J.R.; Berthiaume, L.G. Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 22, 721–732.
- Martin, D.D.O.; Vilas, G.L.; Prescher, J.A.; Rajaiah, G.; Falck, J.R.; Bertozzi, C.R.; Berthiaume, L.G. Rapid detection, discovery, and identification of post-translationally myristoylated proteins during apoptosis using a bio-orthogonal azidomyristate analog. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 22, 797–806.
- Tsiatsiani, L.; Heck, A.J. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626.
- Guo, X.; Trudgian, D.C.; Lemoff, A.; Yadavalli, S.; Mirzaei, H. Confetti: A multiprotease map of the HeLa proteome for comprehensive proteomics. Mol. Cell. Proteom. 2014, 13, 1573–1584.
- Meyer, J.G.; Kim, S.; Maltby, D.A.; Ghassemian, M.; Bandeira, N.; Komives, E.A. Expanding proteome coverage with orthogonal-specificity α-lytic proteases. Mol. Cell. Proteom. 2014, 13, 823–835.
- Bian, Y.; Ye, M.; Song, C.; Cheng, K.; Wang, C.; Wei, X.; Zhu, J.; Chen, R.; Wang, F.; Zou, H. Improve the coverage for the analysis of phosphoproteome of HeLa cells by a tandem digestion approach. J. Proteome Res. 2012, 11, 2828–2837.
- Huesgen, P.F.; Lange, P.F.; Rogers, L.D.; Solis, N.; Eckhard, U.; Kleifeld, O.; Goulas, T.; Gomis-Ruth, F.X.; Overall, C.M. LysargiNase mirrors trypsin for protein C-terminal and methylation-site identification. Nat. Methods 2014, 12, 55–58.
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; Plevak, M.F.; Melton, L.J., 3rd. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2002, 346, 564–569.
- Rajkumar, S.V.; Kyle, R.A.; Buadi, F.K. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: Implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin. Proc. 2010, 85, 945–948.
- Murray, D.L.; Seningen, J.L.; Dispenzieri, A.; Snyder, M.R.; Kyle, R.A.; Rajkumar, S.V.; Katzmann, J.A. Laboratory Persistence and Clinical Progression of Small Monoclonal Abnormalities. Am. J. Clin. Pathol. 2012, 138, 609–613.
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.-T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603.
- Seckinger, A.; Meiβner, T.E.; Moreaux, J.; Depeweg, D.; Hillengass, J.; Hose, K.; Rème, T.; Rösen-Wolff, A.; Jauch, A.; Schnettler, R.; et al. Clinical and prognostic role of annexin A2 in multiple myeloma. Blood 2012, 120, 1087–1094.
- Manier, S.; Boswell, E.N.; Glavey, S.; Sacco, A.; Maiso, P.; Mishima, Y.; Aljaway, Y.; Banwait, R.; Zhang, W.; Zhang, Y.; et al. Mirna Expression Profiling and Proteomic Analysis Of Circulating Exosomes From Multiple Myeloma Patients. Blood 2013, 122, 3086.
- Mailankody, S.; Devlin, S.M.; Korde, N.; Lendvai, N.; Lesokhin, A.; Landau, H.; Hassoun, H.; Ballagi, A.; Ekman, D.; Chung, D.J.; et al. Proteomic profiling in plasma cell disorders: A feasibility study. Leuk. Lymphoma 2016, 58, 1757–1759.
- Kumar, S.; Witzig, T.E.; Timm, M.; Haug, J.; Wellik, L.; Fonseca, R.; Greipp, P.R.; Rajkumar, S.V. Expression of VEGF and its receptors by myeloma cells. Leukemia 2003, 17, 2025–2031.
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2013, 28, 384–390.
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N. Engl. J. Med. 2007, 356, 2582–2590.
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer 2017, 17, 543–556.
- Lussier, T.; Schoebe, N.; Mai, S. Risk Stratification and Treatment in Smoldering Multiple Myeloma. Cells 2021, 11, 130.
- Zhao, J.; Wang, M.; He, P.; Chen, Y.; Wang, X.; Zhang, M. Identification of glutathione S-transferase π 1 as a prognostic proteomic biomarker for multiple myeloma using proteomic profiling. Oncol. Lett. 2020, 19, 2153–2162.
- Yang, W.T.; Zhang, X.J.; Wang, H.; Yang, G.; Xu, X.F. IgD-λ multiple myeloma accompanying with elevated AFP level: A case report and literature review. Int. J. Clin. Exp. Med. 2018, 11, 5176–5180.
- Sahu, A.; Lambris, J.D. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 2001, 180, 35–48.
- Nilsson, B.; Nilsson Ekdahl, K. The tick-over theory revisited: Is C3 a contact-activated protein? Immunobiology 2012, 217, 1106–1110.
- Wlazlo, N.; van Greevenbroek, M.M.; Ferreira, I.; Jansen, E.J.; Feskens, E.J.; van der Kallen, C.J.; Schalkwijk, C.G.; Bravenboer, B.; Stehouwer, C.D. Low-grade inflammation and insulin resistance independently explain substantial parts of the association between body fat and serum C3: The CODAM study. Metabolism 2012, 61, 1787–1796.
- Van Timmeren, M.M.; Chen, M.; Heeringa, P. Review article: Pathogenic role of complement activation in anti-neutrophil cytoplasmic auto-antibody-associated vasculitis. Nephrology 2009, 14, 16–25.
- Xia, S.; Wang, C.; Postma, E.L.; Yang, Y.; Ni, X.; Zhan, W. Fibronectin 1 promotes migration and invasion of papillary thyroid cancer and predicts papillary thyroid cancer lymph node metastasis. OncoTargets Ther. 2017, 10, 1743–1755.
- Ma, L.J.; Lee, S.W.; Lin, L.C.; Chen, T.J.; Chang, I.W.; Hsu, H.P.; Chang, K.Y.; Huang, H.Y.; Li, C.F. Fibronectin overexpression is associated with latent membrane protein 1 expression and has independent prognostic value for nasopharyngeal carcinoma. Tumor Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 35, 1703–1712.
- Sponziello, M.; Rosignolo, F.; Celano, M.; Maggisano, V.; Pecce, V.; De Rose, R.F.; Lombardo, G.E.; Durante, C.; Filetti, S.; Damante, G.; et al. Fibronectin-1 expression is increased in aggressive thyroid cancer and favors the migration and invasion of cancer cells. Mol. Cell. Endocrinol. 2016, 431, 123–132.
- Gao, W.; Liu, Y.; Qin, R.; Liu, D.; Feng, Q. Silence of fibronectin 1 increases cisplatin sensitivity of non-small cell lung cancer cell line. Biochem. Biophys. Res. Commun. 2016, 476, 35–41.
- Yi, W.; Xiao, E.; Ding, R.; Luo, P.; Yang, Y. High expression of fibronectin is associated with poor prognosis, cell proliferation and malignancy via the NF-κB/p53-apoptosis signaling pathway in colorectal cancer. Oncol. Rep. 2016, 36, 3145–3153.
- Jerhammar, F.; Ceder, R.; Garvin, S.; Grénman, R.; Grafström, R.C.; Roberg, K. Fibronectin 1 is a potential biomarker for radioresistance in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2010, 10, 1244–1251.
- Tancred, T.M.; Belch, A.R.; Reiman, T.; Pilarski, L.M.; Kirshner, J. Altered expression of fibronectin and collagens I and IV in multiple myeloma and monoclonal gammopathy of undetermined significance. J. Histochem. Cytochem. 2008, 57, 239–247.
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375.
- Strange, R.C.; Spiteri, M.A.; Ramachandran, S.; Fryer, A.A. Glutathione-S-transferase family of enzymes. Mutat. Res. 2001, 482, 21–26.
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK signaling by GSTp. EMBO J. 1999, 18, 1321–1334.
- Ding, H.; Liu, W.; Yu, X.; Wang, L.; Shao, L.; Yi, W. Risk association of meningiomas with MTHFR C677T and GSTs polymorphisms: A meta-analysis. Int. J. Clin. Exp. Med. 2014, 7, 3904–3914.
- Tew, K.D.; Manevich, Y.; Grek, C.; Xiong, Y.; Uys, J.; Townsend, D.M. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free. Radic. Biol. Med. 2011, 51, 299–313.
- Emadi, A.; Karp, J.E.; Goodman, A.; Ball, E.D.; Megías-Vericat, J.E.; Montesinos, P.; Herrero, M.J.; Bosó, V.; Martínez-Cuadrón, D.; Poveda, J.L.; et al. The clinically relevant pharmacogenomic changes in acute myelogenous leukemia. Pharmacogenomics 2012, 13, 1257–1269.
- Storti, P.; Donofrio, G.; Marchica, V.; Guasco, D.; Todoerti, K.; Airoldi, I.; Anderson, J.; Bolzoni, M.; Ferri, V.; Martella, E.; et al. Galectin-1 Is Highly Expressed By Myeloma Cells and the Bone Marrow Microenvironment and Its Suppression Delineates a New Therapeutic in Vitro and in Vivo Strategy in Multiple Myeloma. Blood 2014, 124, 3373.
- Munshi, N.C.; Hideshima, T.; Carrasco, D.; Shammas, M.; Auclair, D.; Davies, F.; Mitsiades, N.; Mitsiades, C.; Kim, R.S.; Li, C.; et al. Identification of genes modulated in multiple myeloma using genetically identical twin samples. Blood 2004, 103, 1799–1806.
- Lu, G.; Maeda, H.; Reddy, S.V.; Kurihara, N.; Leach, R.; Anderson, J.L.; Roodman, G.D. Cloning and Characterization of the Annexin II Receptor on Human Marrow Stromal Cells. J. Biol. Chem. 2006, 281, 30542–30550.
- D’Souza, S.; Kurihara, N.; Shiozawa, Y.; Joseph, J.; Taichman, R.; Galson, D.L.; Roodman, G.D. Annexin II interactions with the annexin II receptor enhance multiple myeloma cell adhesion and growth in the bone marrow microenvironment. Blood 2012, 119, 1888–1896.
- Sharma, M.C.; Sharma, M. The role of annexin II in angiogenesis and tumor progression: A potential therapeutic target. Curr. Pharm. Des. 2007, 13, 3568–3575.
- Glavey, S.V.; Naba, A.; Manier, S.; Clauser, K.; Tahri, S.; Park, J.; Reagan, M.R.; Moschetta, M.; Mishima, Y.; Gambella, M.; et al. Proteomic characterization of human multiple myeloma bone marrow extracellular matrix. Leukemia 2017, 31, 2426–2434.
- Menaa, C.; Devlin, R.D.; Reddy, S.V.; Gazitt, Y.; Choi, S.J.; Roodman, G.D. Annexin II increases osteoclast formation by stimulating the proliferation of osteoclast precursors in human marrow cultures. J. Clin. Investig. 1999, 103, 1605–1613.
- Storti, P.; Marchica, V.; Airoldi, I.; Donofrio, G.; Fiorini, E.; Ferri, V.; Guasco, D.; Todoerti, K.; Silbermann, R.; Anderson, J.L.; et al. Galectin-1 suppression delineates a new strategy to inhibit myeloma-induced angiogenesis and tumoral growth in vivo. Leukemia 2016, 30, 2351–2363.
- Andersen, M.N.; Ludvigsen, M.; Abildgaard, N.; Petruskevicius, I.; Hjortebjerg, R.; Bjerre, M.; Honoré, B.; Møller, H.J.; Andersen, N.F. Serum galectin-1 in patients with multiple myeloma: Associations with survival, angiogenesis, and biomarkers of macrophage activation. OncoTargets Ther. 2017, 10, 1977–1982.
- Mansour, A.; Wakkach, A.; Blin-Wakkach, C. Emerging Roles of Osteoclasts in the Modulation of Bone Microenvironment and Immune Suppression in Multiple Myeloma. Front. Immunol. 2017, 8, 954.
- Bhattacharyya, S.; Epstein, J.; Suva, L.J. Biomarkers that discriminate multiple myeloma patients with or without skeletal involvement detected using SELDI-TOF mass spectrometry and statistical and machine learning tools. Dis. Markers 2006, 22, 245–255.
- Dowling, P.; Hayes, C.; Ting, K.R.; Hameed, A.; Meiller, J.; Mitsiades, C.; Anderson, K.C.; Clynes, M.; Clarke, C.; Richardson, P.; et al. Identification of proteins found to be significantly altered when comparing the serum proteome from Multiple Myeloma patients with varying degrees of bone disease. BMC Genom. 2014, 15, 904.
- Slany, A.; Haudek-Prinz, V.; Meshcheryakova, A.; Bileck, A.; Lamm, W.; Zielinski, C.; Gerner, C.; Drach, J. Extracellular matrix remodeling by bone marrow fibroblast-like cells correlates with disease progression in multiple myeloma. J. Proteome Res. 2013, 13, 844–854.
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406.
- Chen, Y.; Quan, L.; Jia, C.; Guo, Y.; Wang, X.; Zhang, Y.; Jin, Y.; Liu, A. Proteomics-Based Approach Reveals the Involvement of SERPINB9 in Recurrent and Relapsed Multiple Myeloma. J. Proteome Res. 2021, 20, 2673–2686.
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520.
- Bird, C.H.; Sutton, V.R.; Sun, J.; Hirst, C.E.; Novak, A.; Kumar, S.; Trapani, J.A.; Bird, P.I. Selective regulation of apoptosis: The cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 1998, 18, 6387–6398.
- Pinkoski, M.J.; Waterhouse, N.J.; Heibein, J.A.; Wolf, B.B.; Kuwana, T.; Goldstein, J.C.; Newmeyer, D.D.; Bleackley, R.C.; Green, D.R. Granzyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. J. Biol. Chem. 2001, 276, 12060–12067.
- Hirst, C.E.; Buzza, M.S.; Bird, C.H.; Warren, H.S.; Cameron, P.U.; Zhang, M.; Ashton-Rickardt, P.G.; Bird, P.I. The intracellular granzyme B inhibitor, proteinase inhibitor 9, is up-regulated during accessory cell maturation and effector cell degranulation, and its overexpression enhances CTL potency. J. Immunol. 2003, 170, 805–815.
- Medema, J.P.; Schuurhuis, D.H.; Rea, D.; van Tongeren, J.; de Jong, J.; Bres, S.A.; Laban, S.; Toes, R.E.; Toebes, M.; Schumacher, T.N.; et al. Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte–induced apoptosis: Differential modulation by T helper type 1 and type 2 cells. J. Exp. Med. 2001, 194, 657–668.
- Kaiserman, D.; Bird, P.I. Control of granzymes by serpins. Cell Death Differ. 2009, 17, 586–595.
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558.
- Łuczak, M.; Kubicki, T.; Rzetelska, Z.; Szczepaniak, T.; Przybyłowicz-Chalecka, A.; Ratajczak, B.; Czerwińska-Rybak, J.; Nowicki, A.; Joks, M.; Jakubowiak, A.; et al. Comparative proteomic profiling of sera from patients with refractory multiple myeloma reveals potential biomarkers predicting response to bortezomib-based therapy. Pol. Arch. Intern. Med. 2017, 127, 392–400.
- Yue, X.; He, D.; Zheng, G.; Yang, Y.; Han, X.; Li, Y.; Zhao, Y.; Wu, W.; Chen, Q.; Zhang, E.; et al. Analysis of High-Risk Extramedullary Relapse Factors in Newly Diagnosed MM Patients. Cancers 2022, 14, 6106.
- Bladé, J.; Beksac, M.; Caers, J.; Jurczyszyn, A.; von Lilienfeld-Toal, M.; Moreau, P.; Rasche, L.; Rosiñol, L.; Usmani, S.Z.; Zamagni, E.; et al. Extramedullary disease in multiple myeloma: A systematic literature review. Blood Cancer J. 2022, 12, 45.
- Zatula, A.; Dikic, A.; Mulder, C.; Sharma, A.; Vågbø, C.B.; Sousa, M.M.L.; Waage, A.; Slupphaug, G. Proteome alterations associated with transformation of multiple myeloma to secondary plasma cell leukemia. Oncotarget 2016, 8, 19427–19442.
- Dunphy, K.; Bazou, D.; Dowling, P.; O’Gorman, P. Proteomic Characterisation of the Plasma Proteome in Extramedullary Multiple Myeloma Identifies Potential Prognostic Biomarkers. Blood 2022, 140, 10058–10059.
- Dunphy, K.; Bazou, D.; Henry, M.; Meleady, P.; Dowling, P.; O’Gorman, P. Characterisation of the Tumour Proteome in Primary Extramedullary Multiple Myeloma Identifies Key Proteins Associated with Transendothelial Migration. Blood 2021, 138, 2665.
- Wallington-Beddoe, C.T.; Sobieraj-Teague, M.; Kuss, B.J.; Pitson, S. Resistance to proteasome inhibitors and other targeted therapies in myeloma. Br. J. Haematol. 2018, 182, 11–28.
- Yang, Y.; Chen, Y.; Saha, M.N.; Chen, J.; Evans, K.; Qiu, L.; Reece, D.; Chen, G.A.; Chang, H. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2014, 29, 715–726.
- Ting, K.R.; Henry, M.; Meiller, J.; Larkin, A.; Clynes, M.; Meleady, P.; Bazou, D.; Dowling, P.; O’Gorman, P. Novel panel of protein biomarkers to predict response to bortezomib-containing induction regimens in multiple myeloma patients. BBA Clin. 2017, 8, 28–34.
- Hsieh, F.Y.; Tengstrand, E.; Pekol, T.M.; Guerciolini, R.; Miwa, G. Elucidation of potential bortezomib response markers in mutliple myeloma patients. J. Pharm. Biomed. Anal. 2009, 49, 115–122.
- Dytfeld, D.; Luczak, M.; Wrobel, T.; Usnarska-Zubkiewicz, L.; Brzezniakiewicz, K.; Jamroziak, K.; Giannopoulos, K.; Przybylowicz-Chalecka, A.; Ratajczak, B.; Czerwinska-Rybak, J.; et al. Comparative proteomic profiling of refractory/relapsed multiple myeloma reveals biomarkers involved in resistance to bortezomib-based therapy. Oncotarget 2016, 7, 56726–56736.
- Frassanito, M.A.; De Veirman, K.; Desantis, V.; Di Marzo, L.; Vergara, D.; Ruggieri, S.; Annese, T.; Nico, B.; Menu, E.; Catacchio, I.; et al. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia 2015, 30, 640–648.
- Jones, D.R.; Wu, Z.; Chauhan, D.; Anderson, K.C.; Peng, J. A nano ultra-performance liquid chromatography–high resolution mass spectrometry approach for global metabolomic profiling and case study on drug-resistant multiple myeloma. Anal. Chem. 2014, 86, 3667–3675.
- Maiso, P.; Huynh, D.; Moschetta, M.; Sacco, A.; Aljawai, Y.; Mishima, Y.; Asara, J.M.; Roccaro, A.M.; Kimmelman, A.C.; Ghobrial, I.M. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res 2015, 75, 2071–2082.
- Ng, Y.L.D.; Ramberger, E.; Bohl, S.R.; Dolnik, A.; Steinebach, C.; Conrad, T.; Müller, S.; Popp, O.; Kull, M.; Haji, M.; et al. Proteomic profiling reveals CDK6 upregulation as a targetable resistance mechanism for lenalidomide in multiple myeloma. Nat. Commun. 2022, 13, 1009.
- Chanukuppa, V.; Paul, D.; Taunk, K.; Chatterjee, T.; Sharma, S.; Kumar, S.; Santra, M.K.; Rapole, S. XPO1 is a critical player for bortezomib resistance in multiple myeloma: A quantitative proteomic approach. J. Proteom. 2019, 209, 103504.
- Van der Watt, P.J.; Maske, C.P.; Hendricks, D.T.; Parker, M.I.; Denny, L.; Govender, D.; Birrer, M.J.; Leaner, V.D. The Karyopherin proteins, Crm1 and Karyopherin β1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int. J. Cancer 2009, 124, 1829–1840.
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999, 18, 1660–1672.
- Nikolaev, A.Y.; Li, M.; Puskas, N.; Qin, J.; Gu, W. Parc: A cytoplasmic anchor for p53. Cell 2003, 112, 29–40.
- Noske, A.; Weichert, W.; Niesporek, S.; Röske, A.; Buckendahl, A.C.; Koch, I.; Sehouli, J.; Dietel, M.; Denkert, C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer 2008, 112, 1733–1743.
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014, 7, 85.
- Muqbil, I.; Kauffman, M.; Shacham, S.; Mohammad, R.; Azmi, A. Understanding XPO1 Target Networks Using Systems Biology and Mathematical Modeling. Curr. Pharm. Des. 2014, 20, 56–65.
- Baek, H.B.; Lombard, A.P.; Libertini, S.J.; Fernandez-Rubio, A.; Vinall, R.; Gandour-Edwards, R.; Nakagawa, R.; Vidallo, K.; Nishida, K.; Siddiqui, S.; et al. XPO1 inhibition by selinexor induces potent cytotoxicity against high grade bladder malignancies. Oncotarget 2018, 9, 34567–34581.
- Tan, D.S.; Bedard, P.L.; Kuruvilla, J.; Siu, L.L.; Razak, A.R. Promising SINEs for embargoing nuclear–cytoplasmic export as an anticancer strategy. Cancer Discov. 2014, 4, 527–537.
- Ho, M.; Bianchi, G.; Anderson, K.C. Proteomics-inspired precision medicine for treating and understanding multiple myeloma. Expert Rev. Precis. Med. Drug Dev. 2020, 5, 67–85.
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade®: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513.
- Hambley, B.; Caimi, P.F.; William, B.M. Bortezomib for the treatment of mantle cell lymphoma: An update. Ther. Adv. Hematol. 2016, 7, 196–208.
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nature 2005, 7, 766–772.
- Voorhees, P.M.; Orlowski, R.Z. The proteasome and proteasome inhibitors in cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 189–213.
- Loke, C.; Mollee, P.; McPherson, I.; Walpole, E.; Yue, M.; Mutsando, H.; Wong, P.; Weston, H.; Tomlinson, R.; Hollingworth, S. Bortezomib use and outcomes for the treatment of multiple myeloma. Intern. Med. J. 2020, 50, 1059–1066.
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res 2007, 67, 6383–6391.
- Kuhn, D.J.; Chen, Q.; Voorhees, P.M.; Strader, J.S.; Shenk, K.D.; Sun, C.M.; Demo, S.D.; Bennett, M.K.; van Leeuwen, F.W.B.; Chanan-Khan, A.A.; et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290.
- Groll, M.; Kim, K.B.; Kairies, N.; Huber, R.; Crews, C.M. Crystal Structure of Epoxomicin:20S Proteasome Reveals a Molecular Basis for Selectivity of α‘,β‘-Epoxyketone Proteasome Inhibitors. J. Am. Chem. Soc. 2000, 122, 1237–1238.
- Badros, A.Z.; Vij, R.; Martin, T.; Zonder, J.A.; Kunkel, L.; Wang, Z.; Lee, S.; Wong, A.F.; Niesvizky, R. Carfilzomib in multiple myeloma patients with renal impairment: Pharmacokinetics and safety. Leukemia 2013, 27, 1707–1714.
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J.; et al. U.S. Food and drug administration approval: Carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 2013, 19, 4559–4563.
- Crawford, L.J.; Irvine, A.E. Targeting the ubiquitin proteasome system in haematological malignancies. Blood Rev. 2013, 27, 297–304.
- Brayer, J.; Baz, R. The potential of ixazomib, a second-generation proteasome inhibitor, in the treatment of multiple myeloma. Ther. Adv. Hematol. 2017, 8, 209–220.
- Minarik, J.; Pika, T.; Radocha, J.; Jungova, A.; Straub, J.; Jelinek, T.; Pour, L.; Pavlicek, P.; Mistrik, M.; Brozova, L.; et al. Survival benefit of ixazomib, lenalidomide and dexamethasone (IRD) over lenalidomide and dexamethasone (Rd) in relapsed and refractory multiple myeloma patients in routine clinical practice. BMC Cancer 2021, 21, 73.
- Saltarella, I.; Desantis, V.; Melaccio, A.; Solimando, A.G.; Lamanuzzi, A.; Ria, R.; Storlazzi, C.T.; Mariggiò, M.A.; Vacca, A.; Frassanito, M.A. Mechanisms of Resistance to Anti-CD38 Daratumumab in Multiple Myeloma. Cells 2020, 9, 167.
- Touzeau, C.; Moreau, P. Daratumumab for the treatment of multiple myeloma. Expert Opin. Biol. Ther. 2017, 17, 887–893.
- Zanwar, S.; Nandakumar, B.; Kumar, S. Immune-based therapies in the management of multiple myeloma. Blood Cancer J. 2020, 10, 84.
- Mateos, M.-V.; Sonneveld, P.; Hungria, V.; Nooka, A.K.; Estell, J.A.; Barreto, W.; Corradini, P.; Min, C.-K.; Medvedova, E.; Weisel, K.; et al. Daratumumab, Bortezomib, and Dexamethasone Versus Bortezomib and Dexamethasone in Patients With Previously Treated Multiple Myeloma: Three-year Follow-up of CASTOR. Clin. Lymphoma Myeloma Leuk. 2019, 20, 509–518.
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331.
- Facon, T.; Kumar, S.K.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): Overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 1582–1596.
- Magen, H.; Muchtar, E. Elotuzumab: The first approved monoclonal antibody for multiple myeloma treatment. Ther. Adv. Hematol. 2016, 7, 187–195.
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849.
- Parrondo, R.D.; Paulus, A.; Ailawadhi, S. Updates in the Use of BCL-2-Family Small Molecule Inhibitors for the Treatment of Relapsed/Refractory Multiple Myeloma. Cancers 2022, 14, 3330.
- Ehsan, H.; Wahab, A.; Shah, Z.; Sana, M.K.; Masood, A.; Rafae, A.; Hashmi, H. Role of Venetoclax in the Treatment of Relapsed and Refractory Multiple Myeloma. J. Hematol. 2021, 10, 89–97.


