 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Salla Keskitalo | -- | 4706 | 2023-07-31 13:56:59 | | | |
| 2 | Peter Tang | + 1 word(s) | 4707 | 2023-08-01 05:16:47 | | |
Video Upload Options
CMGC kinases, encompassing cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAPKs), glycogen synthase kinases (GSKs), and CDC-like kinases (CLKs), play pivotal roles in cellular signaling pathways, including cell cycle regulation, proliferation, differentiation, apoptosis, and gene expression regulation. The dysregulation and aberrant activation of these kinases have been implicated in cancer development and progression, making them attractive therapeutic targets. Kinase inhibitors targeting CMGC kinases, such as CDK4/6 inhibitors and BRAF/MEK inhibitors, have demonstrated clinical success in treating specific cancer types.
1. Introduction
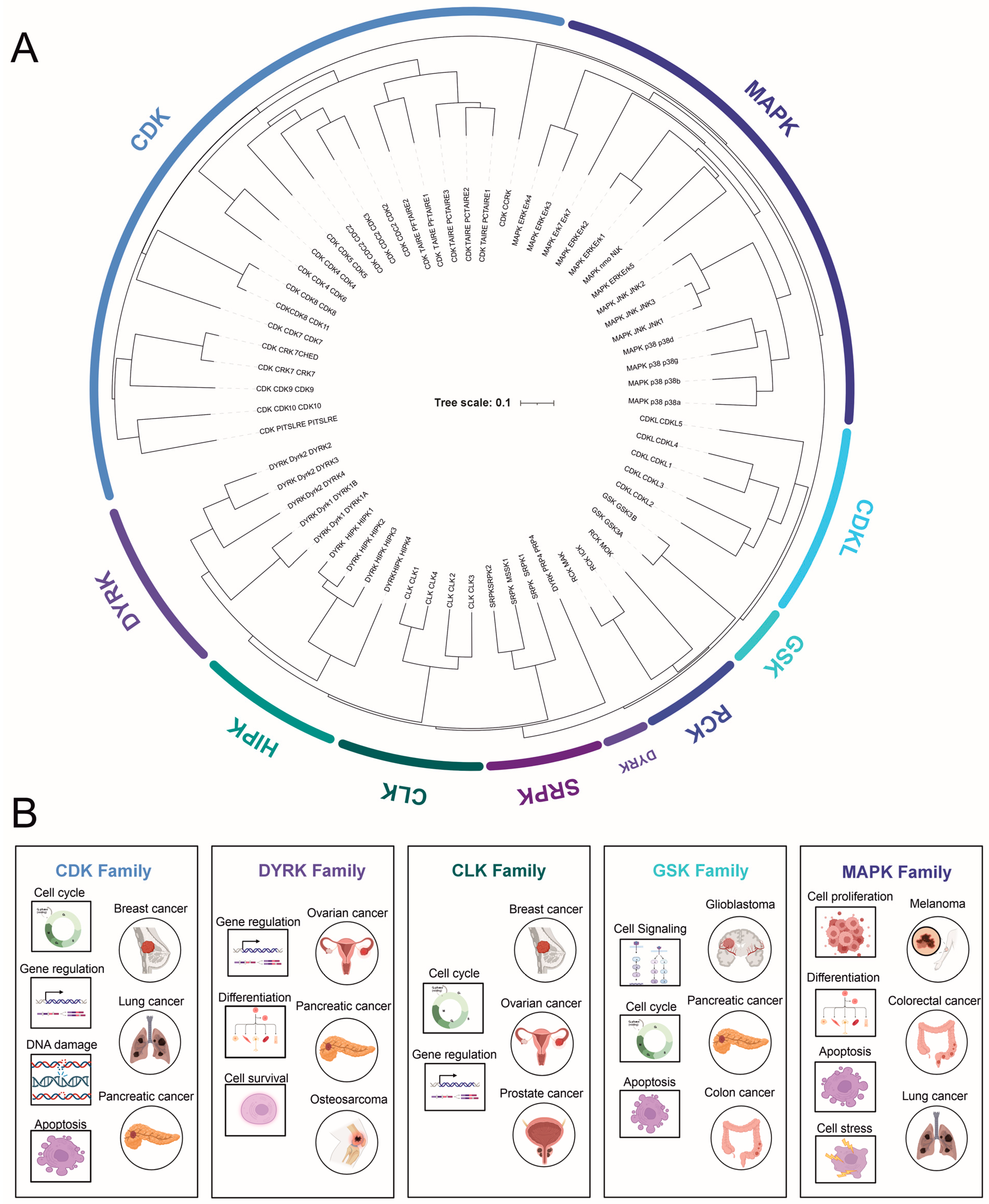
2. CMGC Kinase Subfamilies
2.1. Cyclin-Dependent Kinases (CDKs)
2.2. Mitogen-Activated Protein Kinases (MAPKs)
-
c-Jun N-terminal kinases are highly homologous (>85%) but have a distinct tissue distribution. JNK1 and 2 are ubiquitously expressed, while JNK3 expression is mainly limited to the brain. In contrast to ERKs, JNKs are primarily activated by stress signals such as oxidative stress, radiation, and DNA-damaging agents. JNKs are mainly localized in the cytoplasm, but their identified substrates are mostly TFs, including c-Jun, p53, STAT3, and c-Myc. The phosphorylation of c-Jun leads to AP-1 complex formation and thus the transcription of cyclin D1, promoting cell cycle progression, similar to ERK1/2 [23]. To date, only a few cytoplasmic interaction partners of JNKs have been identified [19][24].
-
The p38 subfamily consists of four members (α, β, γ, δ), which respond to various environmental stress stimuli and cytokines such as interleukin-1 and tumor necrosis factor α(TNF). Interestingly, p38 both regulates the production of cytokines and responds to them. Other targets of p38 regulation are TFs and other protein kinases. Based on observations of p38 activation, it plays a role in inflammation, cell cycle regulation, and apoptosis [19][25].
-
ERK5 (BMK1 or big MAP kinase 1) has a kinase domain similar to ERK1/2, sharing 51% similarity with ERK2. ERK5 is essential during normal embryogenesis [26]. An upstream activator of ERK5 is MEK5, whose expression is elevated in metastatic prostate cancer [27]. Similar to ERK1/2 and JNK, ERK5 also promotes cyclin D1 expression and cell cycle progression [28], as well as plays a crucial role in the maintenance of mitochondrial function and neuronal survival [29]. ERK5 is involved in various cellular processes, including cell survival, differentiation, and angiogenesis. Its activation has been linked to growth factors, oxidative stress, and other extracellular stimuli.
-
The MAPK pathway has a critical role in cancer biology, extending beyond the extensively studied BRAF mutation in melanoma. The MAPK/ERK pathway, for instance, has been implicated in colorectal cancer, with mutations in KRAS and NRAS genes leading to its persistent activation, promoting uncontrolled cell proliferation and tumor growth [30]. These mutations, unfortunately, render the tumors resistant to EGFR-targeted therapies, highlighting the need for novel therapeutic strategies [31].
-
Additionally, the JNK MAPK pathway, associated primarily with responses to stress signals and apoptosis, has shown links to cancer biology. Aberrations in JNK signaling can lead to an imbalance between cell proliferation and death, thereby contributing to oncogenesis. For instance, overactive JNK signaling has been found in several cancers, including breast and gastric cancer, often correlating with a worse prognosis [32]. Furthermore, the p38 MAPK pathway, typically associated with inflammation and cell differentiation, is also relevant in cancer research. Its complex, dual role in tumorigenesis is being unraveled; while its activation can suppress tumor growth by promoting cell cycle arrest and apoptosis, chronic activation can also enhance cancer cell survival, contributing to chemoresistance [33].
2.3. Glycogen Synthase Kinase-3 (GSK-3)
2.4. Dual-Specificity Tyrosine (Y)-Phosphorylation-Regulated Kinases (DYRKs)
2.5. Cdc2-like Kinase (CLK) and Other Less-Studied Kinases
2.6. SR-Specific Protein Kinase (SRPK)
2.7. Tyrosine Kinase Gene v-Ros Cross-Hybridizing Kinase (RCK)
-
MAK (male germ cell-associated kinase) is mainly expressed in testicular germ cells during spermatogenesis and in the retina. In the retina, MAK localizes to connecting cilia in photoreceptor cells, negatively regulates the length of their cilia, and is essential for the survival of these cells [62]. Not surprisingly, MAK mutations are associated with retinitis pigmentosa, a photoreceptor degeneration disease in the retina [63][64].
-
ICK (intestinal cell kinase) is highly conserved, constitutively, and widely expressed. Similarly to MAK, it negatively regulates ciliary length and is identified as an essential component of sonic hedgehog signaling [65]. These two factors seem to be the underlying cause of human ECO syndrome, a multi-organ illness affecting the endocrine, cerebral, and skeletal systems, caused by a missense mutation in the ICK gene [66]. In 2017, a study reported the role of ICK in colorectal cancer progression and its potential as a therapeutic target for the treatment of colorectal cancer [67].
2.8. Cyclin-Dependent Kinase-like (CDKL)
3. Protein Kinase Therapeutics—Kinase Inhibitors
|
Kinase Family |
Deregulation Mechanism |
Examples of Cancer Types |
|---|---|---|
|
CDKs |
Mutations, amplifications, deletions, and altered expression levels |
Breast cancer [77][78][79], lung cancer [80], pancreatic cancer [81] |
|
MAPKs |
Mutations in pathway components (e.g., RAS, RAF) |
Melanoma [82][83][84], colorectal cancer [85], lung cancer [17][86] |
|
DYRK |
Overexpression |
Pancreatic [87][88], ovarian cancer [89][90], osteosarcoma [91], rhabdomyosarcoma [92] |
|
GSKs |
Altered expression levels, post-translational modifications |
Glioblastoma [93], pancreatic cancer [94], colon cancer [95] |
|
CLKs |
Overexpression, cancer-associated splicing alterations |
Breast cancer [96], prostate cancer [97], ovarian cancer [98] |
4. Protein–Protein Interactions of the CMGC Kinases
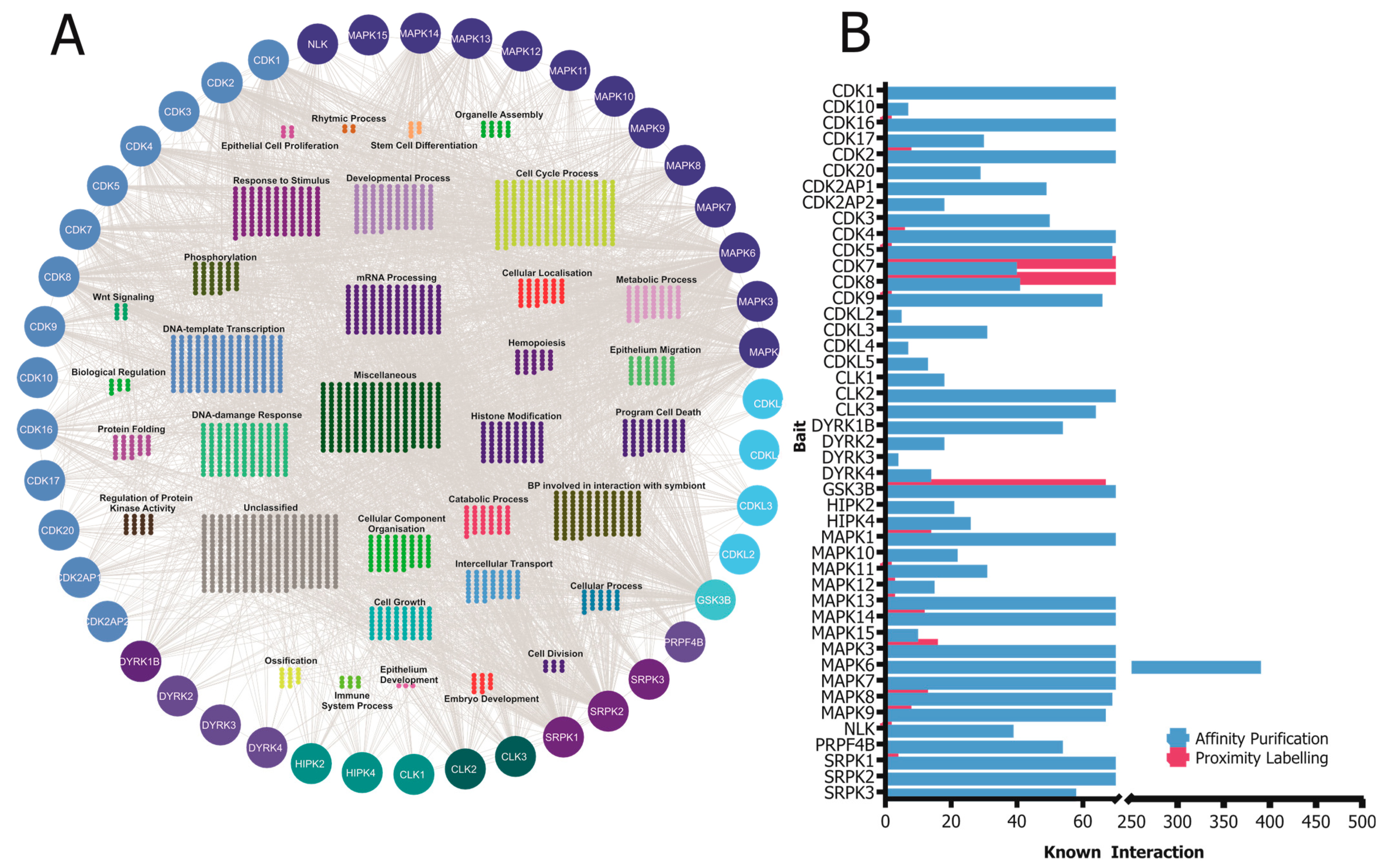
5. Assessment of Kinase Activity
References
- Mann, M.; Ong, S.-E.; Grønborg, M.; Steen, H.; Jensen, O.N.; Pandey, A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002, 20, 261–268.
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77.
- Kannan, N.; Neuwald, A.F. Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2α. Protein Sci. 2004, 13, 2059–2077.
- Nolen, B.; Taylor, S.; Ghosh, G. Regulation of Protein Kinases: Controlling Activity through Activation Segment Conformation. Mol. Cell 2004, 15, 661–675.
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934.
- Pawson, T.; Nash, P. Assembly of Cell Regulatory Systems Through Protein Interaction Domains. Science 2003, 300, 445–452.
- Johnson, L.N.; Noble, M.E.; Owen, D.J. Active and Inactive Protein Kinases: Structural Basis for Regulation. Cell 1996, 85, 149–158.
- Haar, E. Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 2001, 8, 593.
- Varjosalo, M.; Björklund, M.; Cheng, F.; Syvänen, H.; Kivioja, T.; Kilpinen, S.; Sun, Z.; Kallioniemi, O.; Stunnenberg, H.G.; He, W.-W.; et al. Application of Active and Kinase-Deficient Kinome Collection for Identification of Kinases Regulating Hedgehog Signaling. Cell 2008, 133, 537–548.
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.-H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276.
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122.
- Jeffrey, P.D. Mechanism of CDK Activation Revealed by the Structure of a CyclinA-CDK2 Complex. Nature 1995, 376, 313–320.
- Russo, A.A.; Jeffrey, P.D.; Pavletich, N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996, 3, 696–700.
- Harper, J.; Brooks, G. The Mammalian Cell Cycle—An Overwiew. From Methods in Molecular Biology. In Cell Cycle Control: Mechanisms and Protocols; Brooks, T.H., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2005; Volume 296.
- Coulombe, P.; Meloche, S. Atypical mitogen-activated protein kinases: Structure, regulation and functions. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2007, 1773, 1376–1387.
- Deleris, A.; Stroud, H.; Bernatavichute, Y.; Johnson, E.; Klein, G.; Schubert, D.; Jacobsen, S.E. Loss of the DNA Methyltransferase MET1 Induces H3K9 Hypermethylation at PcG Target Genes and Redistribution of H3K27 Trimethylation to Transposons in Arabidopsis thaliana. PLoS Genet. 2012, 8, e1003062.
- Long, W.; Foulds, C.E.; Qin, J.; Liu, J.; Ding, C.; Lonard, D.M.; Solis, L.M.; Wistuba, I.I.; Qin, J.; Tsai, S.Y.; et al. ERK3 signals through SRC-3 coactivator to promote human lung cancer cell invasion. J. Clin. Investig. 2012, 122, 1869–1880.
- Kostenko, S.; Dumitriu, G.; Lægreid, K.J.; Moens, U. Physiological roles of mitogen-activated-protein-kinase-activated p38-regulated/activated protein kinase. World J. Biol. Chem. 2011, 2, 73–89.
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83.
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and p38 Protein Kinases. Science 2002, 298, 1911–1912.
- Burotto, M.; Chiou, V.L.; Lee, J.-M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456.
- Murphy, L.O.; Smith, S.; Chen, R.-H.; Fingar, D.C.; Blenis, J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556–564.
- Sabapathy, K.; Hochedlinger, K.; Nam, S.Y.; Bauer, A.; Karin, M.; Wagner, E.F. Distinct Roles for JNK1 and JNK2 in Regulating JNK Activity and c-Jun-Dependent Cell Proliferation. Mol. Cell 2004, 15, 713–725.
- Bogoyevitch, M.A.; Ngoei, K.R.; Zhao, T.T.; Yeap, Y.Y.; Ng, D.C. c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2010, 1804, 463–475.
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417.
- Regan, C.P.; Li, W.; Boucher, D.M.; Spatz, S.; Su, M.S.; Kuida, K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. USA 2002, 99, 9248–9253.
- Mehta, P.B.; Jenkins, B.L.; McCarthy, L.; Thilak, L.; Robson, C.N.; Neal, D.E.; Leung, H.Y. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene 2003, 22, 1381–1389.
- Mulloy, R.; Salinas, S.; Philips, A.; Hipskind, R.A. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene 2003, 22, 5387–5398.
- Jo, M.; Lee, S.; Kim, K.; Lee, S.; Kim, S.R.; Kim, H.-J. Inhibition of MEK5 suppresses TDP-43 toxicity via the mTOR-independent activation of the autophagy-lysosome pathway. Biochem. Biophys. Res. Commun. 2019, 513, 925–932.
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Origins of Cancer: Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460.
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chien, C.-R.C.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 360, 1408–1417.
- Min, L.; He, B.; Hui, L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin. Cancer Biol. 2011, 21, 10–20.
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549.
- Woodgett, J.R. Molecular Cloning and Expression of Glycogen Synthase Kinase-3/Factor A. EMBO J. 1990, 9, 2431.
- Hoeflich, K.P. Requirement for Glycogen Synthase Kinase-3beta in Cell Survival and NF-KappaB Activation. Nature 2000, 406, 86–90.
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen Synthase Kinase 3α-Specific Regulation of Murine Hepatic Glycogen Metabolism. Cell Metab. 2007, 6, 329–337.
- Cole, A.R.; Knebel, A.; Morrice, N.A.; Robertson, L.A.; Irving, A.J.; Connolly, C.N.; Sutherland, C. GSK-3 Phosphorylation of the Alzheimer Epitope within Collapsin Response Mediator Proteins Regulates Axon Elongation in Primary Neurons. J. Biol. Chem. 2004, 279, 50176–50180.
- Frame, S.; Cohen, P. GSK3 Takes Centre Stage More than 20 Years after Its Discovery. Biochem. J. 2001, 359, 1–16.
- McCubrey, J.A. Effects of Mutations in Wnt/Beta-Catenin, Hedgehog, Notch and PI3K Pathways on GSK-3 Activity-Diverse Effects on Cell Growth, Metabolism and Cancer. Biochim. Biophys. Acta 2016, 1863, 2942.
- Medina, M.; Wandosell, F. Deconstructing GSK-3: The Fine Regulation of Its Activity. Int. J. Alzheimer’s Dis. 2011, 2011, 479249.
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000Research 2017, 6, 167.
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779.
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40.
- Eldar-Finkelman, H.; Krebs, E.G. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc. Natl. Acad. Sci. USA 1997, 94, 9660–9664.
- Ring, D.B.; Johnson, K.W.; Henriksen, E.J.; Nuss, J.M.; Goff, D.; Kinnick, T.R.; Ma, S.T.; Reeder, J.W.; Samuels, I.; Slabiak, T.; et al. Selective Glycogen Synthase Kinase 3 Inhibitors Potentiate Insulin Activation of Glucose Transport and Utilization In Vitro and In Vivo. Diabetes 2003, 52, 588–595.
- Nayler, O.; Stamm, S.; Ullrich, A. Characterization and comparison of four serine- and arginine-rich (SR) protein kinases. Biochem. J. 1997, 326, 693–700.
- Bullock, A.N.; Das, S.; Debreczeni, J.; Rellos, P.; Fedorov, O.; Niesen, F.H.; Guo, K.; Papagrigoriou, E.; Amos, A.L.; Cho, S.; et al. Kinase Domain Insertions Define Distinct Roles of CLK Kinases in SR Protein Phosphorylation. Structure 2009, 17, 352–362.
- Prasad, J.; Colwill, K.; Pawson, T.; Manley, J.L. The Protein Kinase Clk/Sty Directly Modulates SR Protein Activity: Both Hyper- and Hypophosphorylation Inhibit Splicing. Mol. Cell. Biol. 1999, 19, 6991–7000.
- Petsalaki, E.; Zachos, G. Clks 1, 2 and 4 prevent chromatin breakage by regulating the Aurora B-dependent abscission checkpoint. Nat. Commun. 2016, 7, 11451.
- Murai, A.; Ebara, S.; Sasaki, S.; Ohashi, T.; Miyazaki, T.; Nomura, T.; Araki, S. Synergistic apoptotic effects in cancer cells by the combination of CLK and Bcl-2 family inhibitors. PLoS ONE 2020, 15, e0240718.
- Ghosh, G.; Adams, J.A. Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 2010, 278, 587–597.
- Giannakouros, T.; Nikolakaki, E.; Mylonis, I.; Georgatsou, E. Serine-arginine protein kinases: A small protein kinase family with a large cellular presence. FEBS J. 2011, 278, 570–586.
- Calarco, J.A.; Superina, S.; O’Hanlon, D.; Gabut, M.; Raj, B.; Pan, Q.; Skalska, U.; Clarke, L.; Gelinas, D.; van der Kooy, D.; et al. Regulation of Vertebrate Nervous System Alternative Splicing and Development by an SR-Related Protein. Cell 2009, 138, 898–910.
- Hayes, G.M.; Carrigan, P.E.; Miller, L.J. Serine-Arginine Protein Kinase 1 Overexpression Is Associated with Tumorigenic Imbalance in Mitogen-Activated Protein Kinase Pathways in Breast, Colonic, and Pancreatic Carcinomas. Cancer Res. 2007, 67, 2072–2080.
- Hishizawa, M.; Imada, K.; Sakai, T.; Ueda, M.; Hori, T.; Uchiyama, T. Serological identification of adult T-cell leukaemia-associated antigens. Br. J. Haematol. 2005, 130, 382–390.
- Chandra, A.; Ananda, H.; Singh, N.; Qamar, I. Identification of a novel and potent small molecule inhibitor of SRPK1: Mechanism of dual inhibition of SRPK1 for the inhibition of cancer progression. Aging 2020, 13, 163–180.
- Jang, S.-W.; Yang, S.-J.; Ehlén, A.; Dong, S.; Khoury, H.; Chen, J.; Persson, J.L.; Ye, K. Serine/Arginine Protein–Specific Kinase 2 Promotes Leukemia Cell Proliferation by Phosphorylating Acinus and Regulating Cyclin A1. Cancer Res. 2008, 68, 4559–4570.
- Fu, Z.; Schroeder, M.J.; Shabanowitz, J.; Kaldis, P.; Togawa, K.; Rustgi, A.K.; Hunt, D.F.; Sturgill, T.W. Activation of a Nuclear Cdc2-Related Kinase within a Mitogen-Activated Protein Kinase-Like TDY Motif by Autophosphorylation and Cyclin-Dependent Protein Kinase-Activating Kinase. Mol. Cell. Biol. 2005, 25, 6047–6064.
- Miyata, Y.; Akashi, M.; Nishida, E. Molecular cloning and characterization of a novel member of the MAP kinase superfamily. Genes Cells 1999, 4, 299–309.
- Wang, L.-Y.; Kung, H.-J. Male germ cell-associated kinase is overexpressed in prostate cancer cells and causes mitotic defects via deregulation of APC/CCDH1. Oncogene 2011, 31, 2907–2918.
- Fu, Z.; Larson, K.A.; Chitta, R.K.; Parker, S.A.; Turk, B.E.; Lawrence, M.W.; Kaldis, P.; Galaktionov, K.; Cohn, S.M.; Shabanowitz, J.; et al. Identification of Yin-Yang Regulators and a Phosphorylation Consensus for Male Germ Cell-Associated Kinase (MAK)-Related Kinase. Mol. Cell. Biol. 2006, 26, 8639–8654.
- Omori, Y.; Chaya, T.; Katoh, K.; Kajimura, N.; Sato, S.; Muraoka, K.; Ueno, S.; Koyasu, T.; Kondo, M.; Furukawa, T. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc. Natl. Acad. Sci. USA 2010, 107, 22671–22676.
- Özgül, R.K.; Siemiatkowska, A.M.; Yücel, D.; Myers, C.A.; Collin, R.W.; Zonneveld, M.N.; Beryozkin, A.; Banin, E.; Hoyng, C.B.; Born, L.I.v.D.; et al. Exome Sequencing and cis-Regulatory Mapping Identify Mutations in MAK, a Gene Encoding a Regulator of Ciliary Length, as a Cause of Retinitis Pigmentosa. Am. J. Hum. Genet. 2011, 89, 253–264.
- Tucker, B.A.; Scheetz, T.E.; Mullins, R.F.; DeLuca, A.P.; Hoffmann, J.M.; Johnston, R.M.; Jacobson, S.G.; Sheffield, V.C.; Stone, E.M. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2011, 108, E569–E576.
- Moon, H.; Song, J.; Shin, J.-O.; Lee, H.; Kim, H.-K.; Eggenschwiller, J.T.; Bok, J.; Ko, H.W. Intestinal cell kinase, a protein associated with endocrine-cerebro-osteodysplasia syndrome, is a key regulator of cilia length and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 8541–8546.
- Lahiry, P. A multiplex human syndrome implicates a key role for intestinal cell kinase in development of central nervous, skeletal, and endocrine systems. Am. J. Hum. Genet. 2009, 84, 134.
- Chen, T.-M.; Huang, Y.-T.; Wang, G.-C. Outcome of colon cancer initially presenting as colon perforation and obstruction. World J. Surg. Oncol. 2017, 15, 164.
- Chen, J.-Y.; Lin, J.-R.; Tsai, F.-C.; Meyer, T. Dosage of Dyrk1a Shifts Cells within a p21-Cyclin D1 Signaling Map to Control the Decision to Enter the Cell Cycle. Mol. Cell 2013, 52, 87–100.
- Lin, C.; Franco, B.; Rosner, M.R. CDKL5/Stk9 Kinase Inactivation Is Associated with Neuronal Developmental Disorders. Hum. Mol. Genet. 2005, 14, 3775–3786.
- Jiang, Z.; Gong, T.; Wei, H. CDKL5 promotes proliferation, migration, and chemotherapeutic drug resistance of glioma cells via activation of the PI3K/AKT signaling pathway. FEBS Open Bio 2020, 10, 268–277.
- Qin, C.; Ren, L.; Ji, M.; Lv, S.; Wei, Y.; Zhu, D.; Lin, Q.; Xu, P.; Chang, W.; Xu, J. CDKL1 promotes tumor proliferation and invasion in colorectal cancer. OncoTargets Ther. 2017, 10, 1613–1624.
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183.
- Cohen, P. Protein kinases–the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309.
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98.
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130–137.
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789.
- Qian, J.-Y.; Gao, J.; Sun, X.; Cao, M.-D.; Shi, L.; Xia, T.-S.; Zhou, W.-B.; Wang, S.; Ding, Q.; Wei, J.-F. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene 2019, 38, 6123–6141.
- Xing, Z.; Wang, X.; Liu, J.; Zhang, M.; Feng, K.; Wang, X. Expression and prognostic value of CDK1, CCNA2, and CCNB1 gene clusters in human breast cancer. J. Int. Med. Res. 2021, 49, 0300060520980647.
- Wang, N.; Zhang, H.; Li, D.; Jiang, C.; Zhao, H.; Teng, Y. Identification of novel biomarkers in breast cancer via integrated bioinformatics analysis and experimental validation. Bioengineered 2021, 12, 12431–12446.
- Tong, W.; Han, T.-C.; Wang, W.; Zhao, J. LncRNA CASC11 Promotes the Development of Lung Cancer through Targeting MicroRNA-302/CDK1 Axis. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 6539–6547.
- Piao, J.; Zhu, L.; Sun, J.; Li, N.; Dong, B.; Yang, Y.; Chen, L. High expression of CDK1 and BUB1 predicts poor prognosis of pancreatic ductal adenocarcinoma. Gene 2019, 701, 15–22.
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703.
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888.
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85.
- Murugan, A.K.; Dong, J.; Xie, J.; Xing, M. MEK1 mutations, but not ERK2 mutations, occur in melanomas and colon carcinomas, but none in thyroid carcinomas. Cell Cycle 2009, 8, 2122–2124.
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88.
- Luna, J.; Boni, J.; Cuatrecasas, M.; Bofill-De Ros, X.; Núñez-Manchón, E.; Gironella, M.; Vaquero, E.C.; Arbones, M.; de la Luna, S.; Fillat, C. DYRK1A modulates c-MET in pancreatic ductal adenocarcinoma to drive tumour growth. Gut 2018, 68, 1465–1476.
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 2016, 8, 833–845.
- Hu, J.; Nakhla, H.; Friedman, E. Transient arrest in a quiescent state allows ovarian cancer cells to survive suboptimal growth conditions and is mediated by both Mirk/dyrk1b and p130/Rb2. Int. J. Cancer 2010, 129, 307–318.
- Thompson, F.H.; Nelson, M.A.; Trent, J.M.; Guan, X.-Y.; Liu, Y.; Yang, J.-M.; Emerson, J.; Adair, L.; Wymer, J.; Balfour, C.; et al. Amplification of 19q13.1–q13.2 sequences in ovarian cancer: G-band, FISH, and molecular studies. Cancer Genet. Cytogenet. 1996, 87, 55–62.
- Mercer, S.E.; Ewton, D.Z.; Shah, S.; Naqvi, A.; Friedman, E. Mirk/Dyrk1b Mediates Cell Survival in Rhabdomyosarcomas. Cancer Res. 2006, 66, 5143–5150.
- Yang, C.; Ji, D.; Weinstein, E.J.; Choy, E.; Hornicek, F.J.; Wood, K.B.; Liu, X.; Mankin, H.; Duan, Z. The kinase Mirk is a potential therapeutic target in osteosarcoma. Carcinogenesis 2009, 31, 552–558.
- Ugolkov, A.; Qiang, W.; Bondarenko, G.; Procissi, D.; Gaisina, I.; James, C.D.; Chandler, J.; Kozikowski, A.; Gunosewoyo, H.; O’Halloran, T.; et al. Combination Treatment with the GSK-3 Inhibitor 9-ING-41 and CCNU Cures Orthotopic Chemoresistant Glioblastoma in Patient-Derived Xenograft Models. Transl. Oncol. 2017, 10, 669–678.
- Ding, L.; Madamsetty, V.S.; Kiers, S.; Alekhina, O.; Ugolkov, A.; Dube, J.; Zhang, Y.; Zhang, J.-S.; Wang, E.; Dutta, S.K.; et al. Glycogen Synthase Kinase-3 Inhibition Sensitizes Pancreatic Cancer Cells to Chemotherapy by Abrogating the TopBP1/ATR-Mediated DNA Damage Response. Clin. Cancer Res. 2019, 25, 6452–6462.
- Kuroki, H.; Anraku, T.; Kazama, A.; Bilim, V.; Tasaki, M.; Schmitt, D.; Mazar, A.P.; Giles, F.J.; Ugolkov, A.; Tomita, Y. 9-ING-41, a small molecule inhibitor of GSK-3beta, potentiates the effects of anticancer therapeutics in bladder cancer. Sci. Rep. 2019, 9, 19977.
- Yoshida, T.; Kim, J.H.; Carver, K.; Su, Y.; Weremowicz, S.; Mulvey, L.; Yamamoto, S.; Brennan, C.; Mei, S.; Long, H.; et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res. 2015, 75, 1516–1526.
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.-I.; Murray, M.V.; Kimura, H.; et al. Manipulation of Alternative Splicing by a Newly Developed Inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254.
- Liu, Y.; Conaway, L.; Bethard, J.R.; Al-Ayoubi, A.M.; Bradley, A.T.; Zheng, H.; Weed, S.A.; Eblen, S.T. Phosphorylation of the alternative mRNA splicing factor 45 (SPF45) by Clk1 regulates its splice site utilization, cell migration and invasion. Nucleic Acids Res. 2013, 41, 4949–4962.
- Bogoyevitch, M.; Fairlie, D.P. A new paradigm for protein kinase inhibition: Blocking phosphorylation without directly targeting ATP binding. Drug Discov. Today 2007, 12, 622–633.
- Bosc, N.; Meyer, C.; Bonnet, P. The use of novel selectivity metrics in kinase research. BMC Bioinform. 2017, 18, 17.
- Ran, X.; Gestwicki, J.E. Inhibitors of protein–protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86.
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368.
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 2014, 21, 1102–1114.
- Hu, L.; Yang, Y.; Zheng, S.; Xu, J.; Ran, T.; Chen, H. Kinase Inhibitor Scaffold Hopping with Deep Learning Approaches. J. Chem. Inf. Model. 2021, 61, 4900–4912.
- Hastie, C.J.; McLauchlan, H.J.; Cohen, P. Assay of protein kinases using radiolabeled ATP: A protocol. Nat. Protoc. 2006, 1, 968–971.
- Mok, J.; Im, H.; Snyder, M. Global identification of protein kinase substrates by protein microarray analysis. Nat. Protoc. 2009, 4, 1820–1827.
- Zhu, H.; Bilgin, M.; Bangham, R.; Hall, D.; Casamayor, A.; Bertone, P.; Lan, N.; Jansen, R.; Bidlingmaier, S.; Houfek, T.; et al. Global Analysis of Protein Activities Using Proteome Chips. Science 2001, 293, 2101–2105.
- Meng, L.; Michaud, G.A.; Merkel, J.S.; Zhou, F.; Huang, J.; Mattoon, D.R.; Schweitzer, B. Protein kinase substrate identification on functional protein arrays. BMC Biotechnol. 2008, 8, 22.
- Fasolo, J.; Sboner, A.; Sun, M.G.; Yu, H.; Chen, R.; Sharon, D.; Kim, P.M.; Gerstein, M.; Snyder, M. Diverse protein kinase interactions identified by protein microarrays reveal novel connections between cellular processes. Genes Dev. 2011, 25, 767–778.


