 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lucia Giuli | -- | 3440 | 2023-07-29 16:20:45 | | | |
| 2 | Lindsay Dong | -1 word(s) | 3439 | 2023-07-31 04:58:38 | | |
Video Upload Options
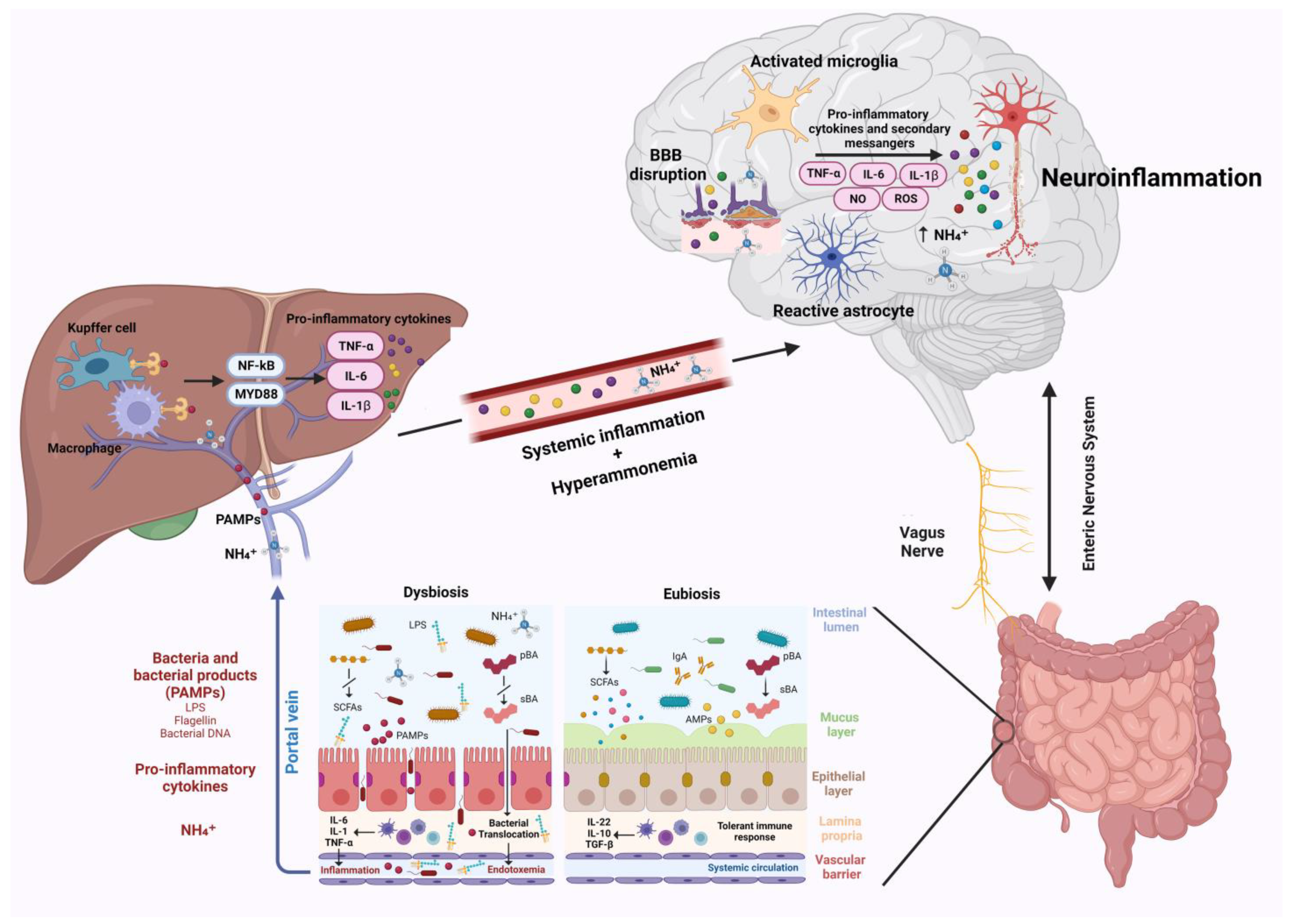
Liver disease are associated with a wide spectrum of neurological changes, of which the best known is hepatic encephalopathy (HE). Historically, hyperammonemia, was considered the main etiological factor; however, studies demonstrated a key role of neuroinflammation in the development of neurological complications in this setting. Neuroinflammation is characterized by activation of microglial cells and brain secretion of pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, which alter neurotransmission, leading to cognitive and motor dysfunction. Changes in the gut microbiota resulting from liver disease play a crucial role in the pathogenesis of neuroinflammation. Dysbiosis and altered intestinal permeability, resulting in bacterial translocation and endotoxemia, are responsible for systemic inflammation, which can spread to brain tissue and trigger neuroinflammation. In addition, metabolites derived from the gut microbiota can act on the central nervous system and facilitate the development of neurological complications, exacerbating clinical manifestations.
1. Introduction
Although HE pathogenic mechanisms are still not fully elucidated, ammonia has always been considered the main causative factor [4]. However, it has been shown that ammonia levels do not correlate with the severity of HE and that HE can also manifest in patients with normal ammonia levels, hinting at the presence of other contributing factors, such as systemic inflammation and oxidative stress [5][6]. Recently several studies have suggested a key role of neuroinflammation in this setting [7]. Indeed, systemic inflammation and hyperammonemia stimulate in concert neuroinflammation [8].
2. Pathophysiology of Neuroinflammation
3. Gut–Liver Axis Contribution to Systemic Inflammation
4. Role of the Gut Microbiota in Hepatic Encephalopathy and Neuroinflammation
5. Neuroinflammation in Acute Liver Failure and Chronic Liver Disease
6. Intestinal Microbiota Modulation as Treatment Strategy and Emerging Therapies
6.1. Rifaximin
6.2. Lactulose
6.3. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
6.4. Fecal Microbiota Transplantation
6.5. Probiotics, Prebiotics and Postbiotics
6.6. Challenges of Proposed Treatments
In conclusion, neuroinflammation appears to be a promising and blooming area of study for the treatment and prevention of HE. The currently available therapeutic strategies appear to be partially effective in modulating neuroinflammation, so it is desirable to identify new effective weapons that are also easily applicable in clinical practice.
References
- 1. Bajaj, J.S. Hepatic Encephalopathy: Classification and Treatment. . J. Hepatol. 2018,, 68, 838–839.
- Montagnese, S.; Rautou, P.-E.; Romero-Gómez, M.; Larsen, F.S.; Shawcross, D.L.; Thabut, D.; Vilstrup, H.; Weissenborn, K. EASL Clinical Practice Guidelines on the Management of Hepatic Encephalopathy. J. Hepatol. 2022, 77, 807–824.
- Amodio, P. Hepatic Encephalopathy: Diagnosis and Management. Liver Int. 2018, 38, 966–975.
- Aldridge, D.R.; Tranah, E.J.; Shawcross, D.L. Pathogenesis of Hepatic Encephalopathy: Role of Ammonia and Systemic Inflammation. J. Clin. Exp. Hepatol. 2015, 5, S7–S20.
- Butterworth, R.F. Hepatic Encephalopathy in Cirrhosis: Pathology and Pathophysiology. . Drugs 2019, 79, 17–21.
- Haj, M.; Rockey, D.C. Ammonia Levels Do Not Guide Clinical Management of Patients with Hepatic Encephalopathy Caused by Cirrhosis. Am. J. Gastroenterol. 2020, 115, 723–728.
- McMillin, M.; DeMorrow, S. Neuroinflammatory Signals during Acute and Chronic Liver Diseases. In Mechanisms of Neuroinflam-mation; Abreu, G.E.A., Ed.; InTech: Houston, TX, USA, 2017.
- Aitbaev, K.A.; Murkamilov, I.T.; Fomin, V.V. Liver Diseases: The Pathogenetic Role of the Gut Microbiome and the Potential of Treatment for Its Modulation. Ter. Arkhiv 2017, 89, 120–128.
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172.
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153.
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate Immune Activation in Neurodegenerative Disease. Nat. Rev. Immunol. 2014, 14, 463–477.
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577.
- Stamatovic, S.; Keep, R.; Andjelkovic, A. Brain Endothelial Cell-Cell Junctions: How to “Open” the Blood Brain Barrier. Curr. Neuropharmacol. 2008, 6, 179–192.
- Sun, Y.; Koyama, Y.; Shimada, S. Inflammation from Peripheral Organs to the Brain: How Does Systemic Inflammation Cause Neuroinflammation? Front. Aging Neurosci. 2022, 14, 903455.
- Salter, M.W.; Stevens, B. Microglia Emerge as Central Players in Brain Disease. Nat. Med. 2017, 23, 1018–1027.
- Tran, V.T.A.; Lee, L.P.; Cho, H. Neuroinflammation in Neurodegeneration via Microbial Infections. Front. Immunol. 2022, 13, 907804.
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science 2005, 308, 1314–1318.
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The Role and Consequences. Neurosci. Res. 2014, 79, 1–12.
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488.
- Eroglu, C.; Barres, B.A. Regulation of Synaptic Connectivity by Glia. Nature 2010, 468, 223–231.
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.-M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A Central Element in Neurological Diseases. Acta Neuropathol. 2016, 131, 323–345.
- Chiareli, R.A.; Carvalho, G.A.; Marques, B.L.; Mota, L.S.; Oliveira-Lima, O.C.; Gomes, R.M.; Birbrair, A.; Gomez, R.S.; Simão, F.; Klempin, F.; et al. The Role of Astrocytes in the Neurorepair Process. Front. Cell Dev. Biol. 2021, 9, 665795.
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622.
- Hoogland, I.C.M.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic Inflammation and Microglial Activation: Systematic Review of Animal Experiments. J. Neuroinflammation 2015, 12, 114.
- Farooq, R.K.; Alamoudi, W.; Alhibshi, A.; Rehman, S.; Sharma, A.R.; Abdulla, F.A. Varied Composition and Underlying Mechanisms of Gut Microbiome in Neuroinflammation. Microorganisms 2022, 10, 705.
- Thursby, E.; Juge, N. Introduction to the Human Gut Microbiota. Biochem. J. 2017, 474, 1823–1836.
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191.
- Baquero, F.; Nombela, C. The Microbiome as a Human Organ. Clin. Microbiol. Infect. 2012, 18, 2–4.
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut Microbiota in Health and Disease. Physiol. Rev. 2010, 90, 859–904.
- Jandhyala, S.M. Role of the Normal Gut Microbiota. World J. Gastroenterol. 2015, 21, 8787.
- Mancini, A.; Campagna, F.; Amodio, P.; Tuohy, K.M. Gut:Liver:Brain Axis: The Microbial Challenge in the Hepatic Encephalopathy. Food Funct. 2018, 9, 1373–1388.
- Jiang, L.; Schnabl, B. Gut Microbiota in Liver Disease: What Do We Know and What Do We Not Know? Physiology 2020, 35, 261–274.
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200.
- Fukiya, S.; Arata, M.; Kawashima, H.; Yoshida, D.; Kaneko, M.; Minamida, K.; Watanabe, J.; Ogura, Y.; Uchida, K.; Itoh, K.; et al. Conversion of Cholic Acid and Chenodeoxycholic Acid into Their 7-Oxo Derivatives by Bacteroides intestinalis AM-1 Isolated from Human Feces. FEMS Microbiol. Lett. 2009, 293, 263–270.
- Trebicka, J.; Macnaughtan, J.; Schnabl, B.; Shawcross, D.L.; Bajaj, J.S. The Microbiota in Cirrhosis and Its Role in Hepatic Decompensation. J. Hepatol. 2021, 75, S67–S81.
- Di Tommaso, N.; Gasbarrini, A.; Ponziani, F.R. Intestinal Barrier in Human Health and Disease. Int. J. Environ. Res. Public Health 2021, 18, 12836.
- Albillos, A.; de Gottardi, A.; Rescigno, M. The Gut-Liver Axis in Liver Disease: Pathophysiological Basis for Therapy. J. Hepatol. 2020, 72, 558–577.
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379.
- Galland, L. The Gut Microbiome and the Brain. J. Med. Food 2014, 17, 1261–1272.
- Rocco, A.; Sgamato, C.; Compare, D.; Coccoli, P.; Nardone, O.M.; Nardone, G. Gut Microbes and Hepatic Encephalopathy: From the Old Concepts to New Perspectives. Front. Cell Dev. Biol. 2021, 9, 748253.
- Bellot, P.; García-Pagán, J.C.; Francés, R.; Abraldes, J.G.; Navasa, M.; Pérez-Mateo, M.; Such, J.; Bosch, J. Bacterial DNA translocation is associated with systemic circulatory abnormalities and intrahepatic endothelial dysfunction in patients with cirrhosis. Hepatology 2010, 52, 2044–2052.
- Cirera, I.; Martin Bauer, T.; Navasa, M.; Vila, J.; Grande, L.; Taurá, P.; Fuster, J.; García-Valdecasas, J.C.; Lacy, A.; Suárez, M.J.; et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 2001, 34, 32–37.
- Muñoz, L.; José Borrero, M.; Ubeda, M.; Lario, M.; Díaz, D.; Francés, R.; Monserrat, J.; Pastor, Ó.; Aguado-Fraile, E.; Such, J.; et al. Interaction between intestinal dendritic cells and bacteria translocated from the gut in rats with cirrhosis. Hepatology 2012, 56, 1861–1869.
- Martin, C.R.; Osadchiy, V.; Kalani, A.; Mayer, E.A. The Brain-Gut-Microbiome Axis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 133–148.
- Foster, J.A.; Neufeld, K.-A.M. Gut–Brain Axis: How the Microbiome Influences Anxiety and Depression. Trends Neurosci. 2013, 36, 305–312.
- El Aidy, S.; Dinan, T.G.; Cryan, J.F. Gut Microbiota: The Conductor in the Orchestra of Immune–Neuroendocrine Communication. Clin. Ther. 2015, 37, 954–967.
- Heiss, C.N.; Olofsson, L.E. The Role of the Gut Microbiota in Development, Function and Disorders of the Central Nervous System and the Enteric Nervous System. J. Neuroendocr. 2019, 31, e12684.
- De Lartigue, G.; de La Serre, C.B.; Raybould, H.E. Vagal Afferent Neurons in High Fat Diet-Induced Obesity; Intestinal Microflora, Gut Inflammation and Cholecystokinin. Physiol. Behav. 2011, 105, 100–105.
- Amodio, P. Hepatic Encephalopathy: Historical Remarks. J. Clin. Exp. Hepatol. 2015, 5, S4–S6.
- Rose, C.F.; Amodio, P.; Bajaj, J.S.; Dhiman, R.K.; Montagnese, S.; Taylor-Robinson, S.D.; Vilstrup, H.; Jalan, R. Hepatic En- cephalopathy: Novel Insights into Classification, Pathophysiology and Therapy. J. Hepatol. 2020, 73, 1526–1547.
- Campion, D.; Giovo, I.; Ponzo, P.; Saracco, G.M.; Balzola, F.; Alessandria, C. Dietary Approach and Gut Microbiota Modulation for Chronic Hepatic Encephalopathy in Cirrhosis. World J. Hepatol. 2019, 11, 489–512.
- Shawcross, D.L.; Sharifi, Y.; Canavan, J.B.; Yeoman, A.D.; Abeles, R.D.; Taylor, N.J.; Auzinger, G.; Bernal, W.; Wendon, J.A. Infection and Systemic Inflammation, Not Ammonia, Are Associated with Grade 3/4 Hepatic Encephalopathy, but Not Mortality in Cirrhosis. J. Hepatol. 2011, 54, 640–649.
- Rai, R.; Saraswat, V.A.; Dhiman, R.K. Gut Microbiota: Its Role in Hepatic Encephalopathy. J. Clin. Exp. Hepatol. 2015, 5, S29–S36.
- Hassouneh, R.; Bajaj, J.S. Gut Microbiota Modulation and Fecal Transplantation: An Overview on Innovative Strategies for Hepatic Encephalopathy Treatment. J. Clin. Med. 2021, 10, 330.
- Chen, Z.; Ruan, J.; Li, D.; Wang, M.; Han, Z.; Qiu, W.; Wu, G. The Role of Intestinal Bacteria and Gut–Brain Axis in Hepatic Encephalopathy. Front. Cell. Infect. Microbiol. 2021, 10, 595759.
- Bajaj, J.S. The Role of Microbiota in Hepatic Encephalopathy. Gut Microbes 2014, 5, 397–403.
- Kawai, T.; Akira, S. Signaling to NF-KB by Toll-like Receptors. Trends Mol. Med. 2007, 13, 460–469.
- Seo, Y.S.; Shah, V.H. The Role of Gut-Liver Axis in the Pathogenesis of Liver Cirrhosis and Portal Hypertension. Clin. Mol. Hepatol. 2012, 18, 337.
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 Enhances TGF-β Signaling and Hepatic Fibrosis. Nat. Med. 2007, 13, 1324–1332.
- Labrenz, F.; Ferri, F.; Wrede, K.; Forsting, M.; Schedlowski, M.; Engler, H.; Elsenbruch, S.; Benson, S.; Costantini, M. Altered Temporal Variance and Functional Connectivity of BOLD Signal Is Associated with State Anxiety during Acute Systemic Inflammation. NeuroImage 2019, 184, 916–924.
- D’Mello, C.; Le, T.; Swain, M.G. Cerebral Microglia Recruit Monocytes into the Brain in Response to Tumor Necrosis Factorα Signaling during Peripheral Organ Inflammation. J. Neurosci. 2009, 29, 2089–2102.
- Chastre, A.; Bélanger, M.; Beauchesne, E.; Nguyen, B.N.; Desjardins, P.; Butterworth, R.F. Inflammatory Cascades Driven by Tumor Necrosis Factor-Alpha Play a Major Role in the Progression of Acute Liver Failure and Its Neurological Complications. PLoS ONE 2012, 7, e49670.
- Frontera, J.A.; Kalb, T. Neurological Management of Fulminant Hepatic Failure. Neurocrit. Care 2011, 14, 318–327.
- Butterworth, R.F. Neuroinflammation in Acute Liver Failure: Mechanisms and Novel Therapeutic Targets. Neurochem. Int. 2011, 59, 830–836.
- Jaeger, V.; DeMorrow, S.; McMillin, M. The Direct Contribution of Astrocytes and Microglia to the Pathogenesis of Hepatic Encephalopathy. J. Clin. Transl. Hepatol. 2019, 7, 352.
- Butterworth, R.F. Pathogenesis of Hepatic Encephalopathy and Brain Edema in Acute Liver Failure. J. Clin. Exp. Hepatol. 2015, 5, S96–S103.
- McMillin, M.; Grant, S.; Frampton, G.; Petrescu, A.D.; Williams, E.; Jefferson, B.; Thomas, A.; Brahmaroutu, A.; DeMorrow, S. Ele- vated Circulating TGFβ1 during Acute Liver Failure Activates TGFβR2 on Cortical Neurons and Exacerbates Neuroinflammation and Hepatic Encephalopathy in Mice. J. Neuroinflammation 2019, 16, 69.
- Cauli, O .; Rodrigo, R.; Piedrafita ,B .; Boix, J.; Felipo, V. Inflammation and Hepatic Encephalopathy: Ibuprofen Restores Learnig Ability in Rats with Portacaval Shunts. Hepatology 2007, 46, 514–519.
- Cagnin , A.In Vivo Imaging of Cerebral “Peripheral Benzodiazepine Binding Sites” in Patients with Hepatic Encephalopathy. Gut 2006, 55, 547–553.
- Ponziani, F.R.; Zocco, M.A.; D’Aversa, F.; Pompili, M.; Gasbarrini, A. Eubiotic Properties of Rifaximin: Disruption of the Traditional Concepts in Gut Microbiota Modulation. World J. Gastroenterol. 2017, 23, 4491.
- Mangas-Losada, A.; García-García, R.;Leone, P.; Ballester, M.P.; Cabrera-Pastor, A.; Urios, A.; Gallego, J.-J.; Martínez-Pretel, J.-J.; Giménez-Garzó, C.; Revert, F.; et al. Selective Improvement by Rifaximin of Changes in the Immunophenotype in Patients Who Improve Minimal Hepatic Encephalopathy. J. Transl. Med. 2019, 17, 293.
- Meng, D.; Yang, M.; Hu, L.; Liu, T.; Zhang, H.; Sun, X.; Wang, X.; Chen, Y.; Jin, Y.; Liu, R. Rifaximin Protects against Circa- dian Rhythm Disruption–Induced Cognitive Impairment through Preventing Gut Barrier Damage and Neuroinflammation. J. Neurochem. 2022, 163, 406–418.
- Li, H.; Xiang, Y.; Zhu, Z.; Wang, W.; Jiang, Z.; Zhao, M.; Cheng, S.; Pan, F.; Liu, D.; Ho, R.C.M. et al. Rifaximin-Mediated Gut Microbiota Regulation Modulates the Function of Microglia and Protects against CUMS-Induced Depression-like Behaviors in Adolescent Rat. J. Neuroinflammation 2021, 18, 254.
- Dalile, B.; VanOudenhove, L.; Vervliet, B.; Verbeke, K. The Role of Short-Chain Fatty Acids in Microbiota–Gut–Brain Communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478.
- Vilstrup, H.; Amodio, P.; Bajaj,J .; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic Encephalopathy in Chronic Liver Disease: 2014 Practice Guideline by the American Association for the Study Of Liver Diseases and the European Association for the Study of the Liver: Vilstrup et Al. Hepatology 2014, 60, 715 735.
- Rodrigo, R.; Cauli, O.; Gomez-Pinedo, U.; Agusti, A.; Hernandez-Rabaza, V.; Garcia-Verdugo, J.; Felipo, V. Hyperammonemia Induces Neuroinflammation That Contributes to Cognitive Impairment in Rats with Hepatic Encephalopathy. Gastroenterology 2010, 139, 675–684.
- Andersson, A. K.; Rönnbäck, L.; Hansson, E. Lactate Induces Tumour Necrosis Factor-α, Interleukin-6 and Interleukin-1β Release in Microglial and Astroglial-Enriched Primary Cultures: Lactate Induces Glial Cell Cytokine Release. J. Neurochem. 2005, 93, 1327–1333.
- Bajaj, J.S.; Ridlon, J.M.; Hylemon, P.B.; Thacker, L.R.; Heuman, D.M.; Smith, S.; Sikaroodi, M.; Gillevet, P.M. Linkage of Gut Microbiome with Cognition in Hepatic Encephalopathy. Am. J. Physiol.—Gastrointest. Liver Physiol. 2012, 302, G168–G175.
- Jia, L. Comparison of Probiotics and Lactulose in the Treatment of Minimal Hepatic Encephalopathy in Rats. WorldJ. Gastroenterol. 2005, 11, 908.
- Jain, L.; Sharma, B.C.; Srivastava, S.; Puri, S.K.; Sharma, P.; Sarin, S. Serum Endotoxin, Inflammatory Mediators, and Magnetic Resonance Spectroscopy before and after Treatment in Patients with Minimal Hepatic Encephalopathy: Inflammatory Mediators in Minimal Hepatic Encephalopathy. J. Gastroenterol. Hepatol. 2013, 28, 1187–1193.
- Elwir, S.; Rahimi, R.S. Hepatic Encephalopathy: An Update on the Pathophysiology and Therapeutic Options .J. Clin. Transl. Hepatol. 2017, 5, 142.
- Bahrami, T.; Yaghmaei, P.; Yousofvand, N. The Effects of Ibuprofen and 1,8 Cineol on Anxiety and Spatial Memory in Hyperammonemic Rats. Metab. Brain Dis. 2023, 38, 613–620.
- Cauli, O.; Rodrigo, R.; Piedrafita, B.; Llansola, M.; Mansouri, M.T.; Felipo, V. Neuroinflammation Contributes to Hypokinesia in Rats with Hepatic Encephalopathy: Ibuprofen Restores Its Motor Activity. J. Neurosci. Res. 2009, 87, 1369–1374.
- Ackerman, Z.; Cominelli, F.; Reynolds, T.B. Effect of Misoprostol on Ibuprofen-Induced Renal Dysfunction in Patients with Decompensated Cirrhosis: Results of a Double-Blind Placebo-Controlled Parallel Group Study. Am. J. Gastroenterol. 2002, 97, 2033–2039.
- Hawkey, C.J. Non-Steroidal Anti-Inflammatory Drug Gastropathy: Causes and Treatment. Scand. J. Gastroenterol .1996,31(Suppl. S220), 124–127.
- Wang, W.-W.; Zhang, Y.;Huang, X.B.; You, N.; Zheng, L.; Li, J. Fecal Microbiota Transplantation Prevents Hepatic Encephalopathy in Rats with Carbon Tetrachloride-Induced Acute Hepatic Dysfunction. World J. Gastroenterol. 2017, 23, 6983–6994.
- Mennigen, R.; Nolte, K.; Rijcken, E.; Utech, M.; Loeffler, B.; Senninger, N.; Bruewer, M. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am. J. Physiol.—Gastrointest. Liver Physiol. 2009, 296, G1140–G1149.
- Albillos, A.; de la Hera, A. Multifactorial gut barrier failure in cirrhosis and bacterial translocation: Working out the role of probiotics and antioxidants. J. Hepatol. 2002, 37, 523–526.
- Ukena, S.N.; Singh, A.; Dringenberg, U.; Engelhardt, R.; Seidler, U.; Hansen, W.; Bleich, A.; Bruder, D.; Franzke, A.; Rogler, G.; et al. Probiotic Escherichia coli Nissle 1917 Inhibits Leaky Gut by Enhancing Mucosal Integrity. PLoS ONE 2007, 2, e1308.
- Dhiman, R.K.; Rana, B.; Agrawal, S.; Garg, A.; Chopra, M.; Thumburu, K.K.; Khattri, A.; Malhotra, S.; Duseja, A.; Chawla,Y.K. Probiotic VSL#3 Reduces Liver Disease Severity and Hospitalization in Patients with Cirrhosis: A Randomized, Controlled Trial. Gastroenterology 2014, 147, 1327–1337.e3.
- Stadlbauer, V.; Mookerjee, R.P.; Hodges, S.; Wright, G.A.K.; Davies, N.A .; Jalan, R. Effect of probiotic treatment on deranged neutrophil function and cytokine responses in patients with compensated alcoholic cirrhosis. J. Hepatol. 2008, 48, 945–951.
- Horvath, A.; Leber, B.; Schmerboeck, B.; Tawdrous, M.; Zettel, G.; Hartl, A.; Madl, T.; Stryeck, S.; Fuchs, D.; Lemesch, S.;et al. Randomised clinical trial: The effects of a multispecies probiotic vs. placebo on innate immune function, bacterial translocation and gut permeability in patients with cirrhosis. Aliment. Pharmacol. Ther. 2016, 44, 926–935.
- Bajaj, J.S.; Heuman, D.M.; Hylemon, P.B.; Sanyal, A.J.; Puri, P.; Sterling, R.K.; Luketic, V.; Stravitz, R.T.; Siddiqui, M.S.; Fuchs, M.; et al. Randomised clinical trial: Lactobacillus GG modulates gut microbiome, metabolome and endotoxemia in patients with cirrhosis. Aliment. Pharmacol. Ther. 2014, 39, 1113–1125.
- Cao, Q.; Yu, C.-B.; Yang,S.-G.;Cao, H.-C.; Chen, P.; Deng, M.; Li,L.-J. Effect of probiotic treatment on cirrhotic patients with minimal hepatic encephalopathy: A meta-analysis. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 9–16.
- Ponziani, F.R. Effect of rifaximin on gut microbiota composition in advanced liver disease and its complications. World J. Gastroenterol. 2015, 21, 12322.
- Hudson, M.; Schuchmann, M. Long-term management of hepatic encephalopathy with lactulose and/or rifaximin: A review of the evidence. Eur. J. Gastroenterol. Hepatol. 2019, 31, 434–450.
- Montagnese, S.; Russo, F.P.; Amodio, P.; Burra, P.; Gasbarrini, A.; Loguercio, C.; Marchesini, G.; Merli, M.; Ponziani, F.R.; Riggio, O.; et al. Hepatic encephalopathy 2018: A clinical practice guideline by the Italian Association for the Study of the Liver (AISF). Dig. Liver Dis. 2019, 51, 190–205.
- Bajaj, J.S.; Riggio, O. Drug therapy: Rifaximin1. Hepatology 2010, 52, 1484–1488.
- Sbahi, H.; Di Palma, J.A. Faecal microbiota transplantation: Applications and limitations in treating gastrointestinal disorders. BMJ Open Gastroenterol. 2016, 3, e000087.
- Shi, Y.-C.; Yang,Y.S. Fecal microbiota transplantation: Current status and challenges in China: FMT in China. JGH Open 2018, 2, 114–116.
- De Filipp, Z.; Bloom, P.P.; Soto, M.T.; Mansour, M.K.; Sater, M.R.A.; Huntley, M.H.; Turbett, S.; Chung, R.T.; Chen,Y.-B.; Hohmann, E.L. Drug-Resistant E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N. Engl. J. Med. 2019, 381, 2043–2050.

