Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Karim Ullah | -- | 2169 | 2023-07-25 14:00:48 | | | |
| 2 | Rita Xu | Meta information modification | 2169 | 2023-07-26 03:57:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ullah, K.; Ai, L.; Humayun, Z.; Wu, R. HIF2α/ARNT Expression for Ischemic Heart Disease Therapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/47253 (accessed on 23 July 2026).
Ullah K, Ai L, Humayun Z, Wu R. HIF2α/ARNT Expression for Ischemic Heart Disease Therapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/47253. Accessed July 23, 2026.
Ullah, Karim, Lizhuo Ai, Zainab Humayun, Rongxue Wu. "HIF2α/ARNT Expression for Ischemic Heart Disease Therapy" Encyclopedia, https://encyclopedia.pub/entry/47253 (accessed July 23, 2026).
Ullah, K., Ai, L., Humayun, Z., & Wu, R. (2023, July 25). HIF2α/ARNT Expression for Ischemic Heart Disease Therapy. In Encyclopedia. https://encyclopedia.pub/entry/47253
Ullah, Karim, et al. "HIF2α/ARNT Expression for Ischemic Heart Disease Therapy." Encyclopedia. Web. 25 July, 2023.
Copy Citation
Ischemic heart disease (IHD) is a major cause of mortality and morbidity worldwide, with novel therapeutic strategies urgently needed. Endothelial dysfunction is a hallmark of IHD, contributing to its development and progression. Hypoxia-inducible factors (HIFs) are transcription factors activated in response to low oxygen levels, playing crucial roles in various pathophysiological processes related to cardiovascular diseases.
ischemic heart disease
endothelial cell function
HIF2α
ARNT
1. Introduction
Ischemic heart disease (IHD) is a prevalent condition resulting from insufficient oxygen supply to the heart muscle, causing significant morbidity and mortality worldwide [1][2][3]. Tissue survival and healthy organ function are dependent on adequate oxygen supply. Oxygen levels may also fluctuate in tissues, depending on the metabolic demand of the tissue and the oxygen tension in the blood. Therefore, local oxygen partial pressure acts as the main functional regulator of oxygen homeostasis. As the first responder to hypoxia, endothelial cells lining the microvasculature activate multiple signaling pathways to compensate for low oxygen tension, such as increased vasodilation and the expression of hypoxia-inducible factors (HIFs) [4][5]. HIFs are transcription factors that play a crucial role in cellular adaptation to hypoxia, and three well-recognized isoforms exist: HIF1α, HIF2α, and HIF3α, which are encoded by three distinct genes [6][7]. HIF1α and HIF2α share a similar domain composition [8], whereas HIF3α has high similarity in the bHLH and PAS domains with HIF1α and HIF2α but lacks the C-terminal transactivation domain (CTAD) [9]. HIF1α and HIF2α are the most well-studied members of this family and share a similar structure. Both are unstable under normal oxygen conditions (normoxia) due to the activity of prolyl hydroxylases (PHDs) that hydroxylate HIF1α and HIF2α, marking them for degradation. Under low oxygen conditions (hypoxia), the activity of PHDs is inhibited, leading to the stabilization of HIF1α and HIF2α. Upon stabilization, both HIF1α and HIF2α translocate to the nucleus, where they each dimerize with the aryl hydrocarbon receptor nuclear translocator (ARNT, also known as HIF1β). The resulting heterodimeric complexes, HIF1 (HIF1α/ARNT) and HIF2 (HIF2α/ARNT), then bind to hypoxia-response elements (HREs) in the promoters of target genes and activate their transcription. Although both HIF1 and HIF2 complexes bind to HREs and regulate gene expression in response to hypoxia, they are not identical in their functions. Hif1α gene transcriptions primarily govern metabolic reprogramming, while Hif2α plays a more significant role in regulating angiogenic extracellular signaling, guidance cues, and factors related to remodeling the extracellular matrix.
2. Role of HIF2α and ARNT in Vascular Endothelial Cells and Inflammation
2.1. HIF2a Expression and Inflammation
HIF2α, also known as endothelial PAS domain protein 1 (EPAS1), is abundantly expressed in vascular endothelial cells. HIF2α is a paralog of HIF1α (48% similarity) that also binds to ARNT [10]. Instead of being expressed in most cell types like HIF1α, the expression of HIF2α is confined to more specific cells and tissues, such as hepatocytes, cardiomyocytes, lungs, kidney interstitial cells, and some cell types in the central nervous system [10][11]. HIF2α binds to the hypoxia response element (HRE) and enhances the expression of genes for erythropoietin, vascular endothelial growth factor (VEGF), and various glycolytic enzymes, as well as activates the transcription of a reporter gene harboring the HRE [12]. Several studies have shown that HIF1α and HIF2α proteins are similarly induced by acute hypoxia in human lungs (4 h, 0.5% O2) at the translational or posttranslational level, but HIF1α protein stimulation disappears because of a reduction in its mRNA stability by prolonged hypoxia (12 h, 0.5% O2), whereas HIF2α protein stimulation remains high and stable during prolonged hypoxia [13]. Hif2α knockout mice experience perinatal deaths, but a small number of surviving Hif2α KO mice exhibit significant hematopoietic defects [14].
Inflammation and hypoxia are recognized as hallmarks of many pathological conditions and have been implicated in the pathogenesis of ischemic heart diseases. The presence of these conditions leads to the stabilization and activation of hypoxia-inducible factors (HIFs) in inflamed cells [15][16]. In a previously described ischemic kidney injury model, the authors found that endothelial Hif2α, but not Hif1α, regulated kidney inflammation via suppressing Vcam1 expression [17]. Ischemia–reperfusion injury (IRI) of the kidneys revealed a significant increase of Vcam1 mRNA expression in endothelial cell-specific Hif2α knockout kidneys, leading to prolonged leukocyte adhesion and transendothelial migration, which further exacerbates inflammation [17]. On the other hand, the Hif2α-dependent induction of amphiregulin expression results in faster recovery from myocardial IRI [18], and amphiregulin is known to suppress local inflammation [19]. Interestingly, global Hif2α knockdown-induced renal injury can be reversed by the restoration of HIF2α in ECs [17].
Inflammatory stimuli such as LPS have been shown to significantly decrease HIF2α expression and increase the infiltration of inflammatory cells into the heart and the lungs. However, HIF2α induction through inactivating PHD2 reverses LPS-induced cardiac dysfunction [20]. Conversely, endothelial Hif2α deletion has been found to increase inflammation and induce lung vascular leakage [21]. It was reported that LPS stimulation enhances the expression of PHD2, leading to a decrease in the levels of Notch3 and HIF2α. However, this reduction in HIF2α and Notch3 expression was reversed by the overexpression of Sirtuin 3 [20]. Mechanistically, LPS stimulation suppresses the expression of Sirtuin-3, HIF2α, and Notch3 while promoting the expression of PHD2 and ang2 (Figure 1). Interestingly, the overexpression of Sirtuin 3 stimulates the expression of Ang-1/Tie-2 while reducing the expression of ang2 [22][23]. Additionally, Sirtuin 3 overexpression induces the expression of HIF2α and Notch3 [22][23]. Based on the above experimental evidence, it suggests that the induction of HIF2α may limit inflammation and promote endothelial cell survival by preserving the barrier integrity (Figure 1). Although the function of HIF2α in tissue inflammation has been studied in multiple organs and disease models, most studies suggest that HIF2α is protective in acute organ injuries but oncogenic during tumor development.
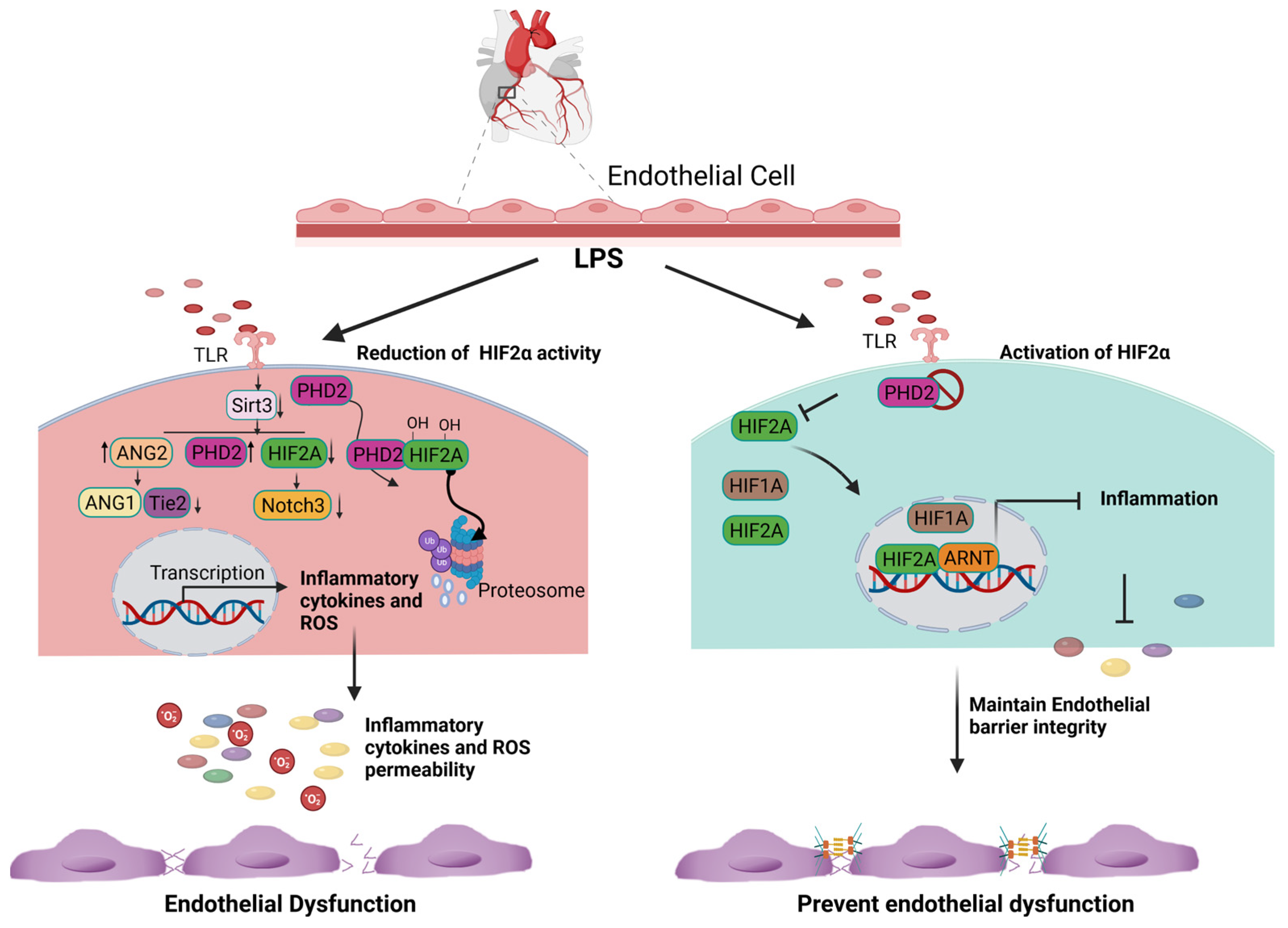
Figure 1. Cardioprotective role of endothelial HIF2A expression. LPS stimulation increases the expression of PHD2 and limits the amount of HIF2A via the hydroxylation-dependent degradation of HIF2A. In the absence of HIF2A, inflammatory genes are transcribed and generate excessive ROS. Consequently, it increases endothelial cell apoptosis and loss of the endothelial barrier function (Right panel). The inhibition of PHD2 results in the accumulation and translocation of HIF2A into the nucleus. HIF2A binds to ARNT, limits the expression of inflammatory genes, and subsequently decreases ROS generation. The inhibition of ROS production and inflammation prevent endothelial barrier dysfunction.
2.2. The Role of ARNT in Heterodimeric Transcription Factors and Endothelial Cell Function
The aryl hydrocarbon receptor nuclear translocator (ARNT), also known as HIF1β, is a transcription factor and belongs to the basic helix–loop–helix Per/Arnt/Sim (bHLH-PAS) superfamily of transcription factors [24]. Both the HIFα and HIF1β (ARNT) proteins share a common structural bHLH domain, which is responsible for dimerization and DNA binding [25]. As a NLS-containing transcription factor, ARNT was first identified as a factor required for the nuclear translocation of the ligand-bound aryl hydrocarbon receptor (AHR) [26]. ARNT is necessary for the generation of heterodimeric transcription factors with HIF-1 and HIF-2 alpha subunits and regulates the expression of genes involved in cell survival, proliferation, angiogenesis, and metabolism under hypoxia conditions [27][28]. For example, in mouse hepatoma cell lines, the induction of HIF target genes depends on the heterodimerization with ARNT [29], and Arnt knockout embryonic stem cells fail to induce the hypoxia-dependent expression of genes [30]. In primary endothelial cells, the loss of ARNT leads to reduced viability of the cells without affecting their proliferation [31].
3. Potential Role of HIF2α and ARNT in Ischemic Heart Disease
3.1. HIF2α and ARNT in Mouse Heart Development and Cardiovascular Function
Mouse heart development occurs between embryonic days E7.75–15, with various cells of different origins contributing to heart development [32]. During embryogenesis, the rapid growth of embryonic tissue increases oxygen consumption, creating a hypoxic microenvironment [33] and activating the components of HIF (HIF1α, HIF2α, and ARNT) [34][35]. Vascular endothelial cells are fundamentally important for heart development and are associated with both surfaces of the myocardium. The myocardium is generally hypoxic during the early stage of development [32][36]. Vascular endothelial cell and myocardium development depend on a reciprocal interaction [37][38]. The loss of HIF activity results in both myocardial and endocardial defects during development. In vivo studies have suggested that Hif1α is essential in the myocardium but not in the endothelium for normal development of the heart [32]. Mice lacking functional Hif2α in vascular endothelial cells develop multiple cardiovascular defects, including a thin myocardium, a disorganized endocardium, and irregular trabeculation [39].
Mouse genetic studies have revealed the critical role of HIF signaling pathways in vascular development and pathogenesis. The genetic inactivation of Hif1α or Arnt results in embryonic lethality due to abnormal vascular development, while the inhibition of Hif2α leads to impairment in vascular remodeling and an altered cardiac rhythm, depending on the genetic background of the mice [40][41]. HIF isoforms are expressed at different heart developmental stages in different cell types [42]. HIF1α is mainly expressed in the cardiomyocyte, while HIF2α is predominantly expressed in endothelial cells in the heart during embryonic development [43]. Hif1α+/− mice develop normally but show impaired physiological responses to chronic hypoxia. One strain of Hif2α-deficient mice displayed hemorrhage and failed to maintain discrete vascular tubular structures, indicating Hif2α’s essential role in vascular remodeling during development.
In addition to embryonic development, Hif2α plays a key role in ischemic heart disease (IHD). IHD is a condition where the blood supply to the heart muscle is reduced, typically due to atherosclerosis or coronary artery blockage, leading to inadequate oxygen supply to the heart tissue [44]. HIF2α expression in vascular endothelial cells during IHD has several functional consequences. For example, myocyte-specific Hif2α deletion enhances ischemic injury in mouse models [18]. One study showed that EC-specific Hif2α deletion in mice impaired angiogenesis in its hindlimb ischemia and autochthonous solid tumor model [45][46]. Under hypoxic conditions, it has been observed that the HIF2α isoform plays a pivotal role in regulating the expression of the DiI4, Adm1, and Ang2 genes [45]. This indicates that Hif2α is critically involved in facilitating complementary angiogenesis within ischemic tissue at a mechanistic level. In addition, HIF2α induces the expression of proangiogenic factors such as vascular endothelial growth factor (VEGF) [47]. The induction of VEGF stimulates the growth of new blood vessels and facilitates blood flow to ischemic tissues [48]. A recent study revealed that the induction of HIF2α promotes endothelial cell survival under hypoxia and maintains the endothelial barrier integrity [21][46][49]. These findings suggest that HIF2A enhances endothelial cell survival under ischemic conditions. In summary, HIF2α is not only critical for embryonic development but also plays a key role in the pathogenesis of ischemic heart disease. Its involvement in promoting angiogenesis, inducing the expression of proangiogenic factors, and enhancing endothelial cell survival under hypoxia highlights its potential as a therapeutic target for the treatment of IHD.
3.2. Cardioprotective Potential of ARNT and HIF2α
ARNT plays a significant role in preventing the development of cardiomyopathy and subsequent heart failure. The cardiac-specific deletion of Arnt in mice leads to PPARα activation, dilated left ventricular chambers, and impaired cardiac contractility, resembling diabetic cardiomyopathy [50]. Furthermore, the endothelial cell-specific deletion of Arnt results in increased cardiomyocyte size and impaired cardiac contractility [51]. ARNT also protects against cardiac, renal, and liver fibrosis by acting through the FKBP-YY1-ARNT-ALK3 signaling pathway [52]. The deletion of Arnt promotes fibrosis by increasing collagen 1A1 and MMP9 production [53]. The inhibition of FKBP12/YY1 increased ARNT expression, reducing organ damage in renal, heart, and liver fibrosis models [54][55]. These findings underscore ARNT’s protective role in the development and progression of heart failure, making it a potential therapeutic target for ischemic heart failure and the treatment for maladaptive cardiac fibrosis in chronic inflammatory heart diseases.
HIF2α is highly expressed in vascular endothelial cells and regulates the expression of target genes responsible for vascular function and angiogenesis [45]. Population genetic and animal model studies suggest that HIF2α is a critical regulator of several cardiovascular diseases [42][50][56]. Indeed, HIF2α plays a crucial role in maintaining vascular integrity. For example, endothelial cell-specific HIF2α deletion increases vascular permeability in multiple organs, including the lungs [45]. In ischemia–reperfusion injury of the kidneys, activation of HIF2α by using PHD inhibitor FG4487 protects against renal failure [57]. Similarly, the use of l-mimosine and dimethyloxalylglycine, which inactivate prolyl-4 hydroxylases, thereby activating HIF signaling, protects mouse kidneys from injury induced by ischemia–reperfusion in the kidneys [58]. HIF is considered a central component of ischemic preconditioning of the heart, in which repetitive short exposure to ischemia and reperfusion is given before a subsequent long exposure to ischemia and helps to preadapt the myocardium to activate protective mechanisms. Several studies have shown the cardioprotective effects of HIF activation during ischemic injury [59]. The induction of HIF2α provides protective mechanisms in cardiomyocytes’ adaptation to chronic hypoxia [60]. Furthermore, the deletion of HIF2α, rather than HIF1α, demonstrates the essential role of HIF2α in myocytes for improving the tolerance to myocardial ischemia–reperfusion injury. This is achieved through the upregulation of its specific target gene, amphiregulin [18]. In contrast, the induction of HIF2α by inactivating PHD2 protects the mouse heart from acute ischemia–reperfusion injury [61]. As HIF2α is highly expressed in endothelial cells, the activation of HIF2α in these cells, either by inactivating HIF2α-degrading enzymes, such as PHD2, or by overexpressing HIF2α, could provide a novel therapeutic approach for the treatment of cardiovascular disease. However, the role of endothelial HIF2α in ischemic heart diseases requires further investigation.
References
- Lopez-Barneo, J.; Pardal, R.; Ortega-Saenz, P. Cellular mechanism of oxygen sensing. Annu. Rev. Physiol. 2001, 63, 259–287.
- Ferdinand, P.; Roffe, C. Hypoxia after stroke: A review of experimental and clinical evidence. Exp. Transl. Stroke Med. 2016, 8, 9.
- Cornet, A.D.; Kooter, A.J.; Peters, M.J.; Smulders, Y.M. The potential harm of oxygen therapy in medical emergencies. Crit. Care 2013, 17, 313.
- Michiels, C.; Arnould, T.; Remacle, J. Endothelial cell responses to hypoxia: Initiation of a cascade of cellular interactions. Biochim. Biophys. Acta 2000, 1497, 1–10.
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669.
- Wu, D.; Rastinejad, F. Structural characterization of mammalian bHLH-PAS transcription factors. Curr. Opin. Struct. Biol. 2017, 43, 1–9.
- Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouyssegur, J. A dialogue between the hypoxia-inducible factor and the tumor microenvironment. Cancer Microenviron. 2008, 1, 53–68.
- Downes, N.L.; Laham-Karam, N.; Kaikkonen, M.U.; Yla-Herttuala, S. Differential but Complementary HIF1α and HIF2α Transcriptional Regulation. Mol. Ther. 2018, 26, 1735–1745.
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and characterization of hypoxia-inducible factor (HIF)-3α in human kidney: Suppression of HIF-mediated gene expression by HIF-3α. Biochem. Biophys. Res. Commun. 2001, 287, 808–813.
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82.
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors—Similar but not identical. Mol. Cells 2010, 29, 435–442.
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278.
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1α and HIF-2α expression in lung epithelial cells: Implication of natural antisense HIF-1α. J. Biol. Chem. 2004, 279, 14871–14878.
- Scortegagna, M.; Morris, M.A.; Oktay, Y.; Bennett, M.; Garcia, J.A. The HIF family member EPAS1/HIF-2α is required for normal hematopoiesis in mice. Blood 2003, 102, 1634–1640.
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665.
- Bartels, K.; Grenz, A.; Eltzschig, H.K. Hypoxia and inflammation are two sides of the same coin. Proc. Natl. Acad. Sci. USA 2013, 110, 18351–18352.
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Invest. 2014, 124, 2396–2409.
- Koeppen, M.; Lee, J.W.; Seo, S.W.; Brodsky, K.S.; Kreth, S.; Yang, I.V.; Buttrick, P.M.; Eckle, T.; Eltzschig, H.K. Hypoxia-inducible factor 2-α-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat. Commun. 2018, 9, 816.
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015, 42, 216–226.
- Zeng, H.; He, X.; Tuo, Q.H.; Liao, D.F.; Zhang, G.Q.; Chen, J.X. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3 pathways. Sci. Rep. 2016, 6, 20931.
- Gong, H.; Rehman, J.; Tang, H.; Wary, K.; Mittal, M.; Chaturvedi, P.; Zhao, Y.Y.; Komarova, Y.A.; Vogel, S.M.; Malik, A.B. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J. Clin. Invest. 2015, 125, 652–664.
- Wei, T.; Gao, J.; Huang, C.; Song, B.; Sun, M.; Shen, W. SIRT3 (Sirtuin-3) Prevents Ang II (Angiotensin II)-Induced Macrophage Metabolic Switch Improving Perivascular Adipose Tissue Function. Arter. Thromb. Vasc. Biol. 2021, 41, 714–730.
- Zhang, C.; Li, N.; Suo, M.; Zhang, C.; Liu, J.; Liu, L.; Qi, Y.; Zheng, X.; Xie, L.; Hu, Y.; et al. Sirtuin 3 deficiency aggravates angiotensin II-induced hypertensive cardiac injury by the impairment of lymphangiogenesis. J. Cell. Mol. Med. 2021, 25, 7760–7771.
- Bersten, D.C.; Sullivan, A.E.; Peet, D.J.; Whitelaw, M.L. bHLH-PAS proteins in cancer. Nat. Rev. Cancer 2013, 13, 827–841.
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Kohler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin α/β pathway. Biochim. Biophys. Acta 2008, 1783, 394–404.
- Hoffman, E.C.; Reyes, H.; Chu, F.F.; Sander, F.; Conley, L.H.; Brooks, B.A.; Hankinson, O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science 1991, 252, 954–958.
- Ullah, K.; Wu, R. Hypoxia-Inducible Factor Regulates Endothelial Metabolism in Cardiovascular Disease. Front. Physiol. 2021, 12, 670653.
- Nguyen, T.; Zheng, M.; Knapp, M.; Sladojevic, N.; Zhang, Q.; Ai, L.; Harrison, D.; Chen, A.; Sitikov, A.; Shen, L.; et al. Endothelial Aryl Hydrocarbon Receptor Nuclear Translocator Mediates the Angiogenic Response to Peripheral Ischemia in Mice With Type 2 Diabetes Mellitus. Front. Cell Dev. Biol. 2021, 9, 691801.
- Wood, S.M.; Gleadle, J.M.; Pugh, C.W.; Hankinson, O.; Ratcliffe, P.J. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J. Biol. Chem. 1996, 271, 15117–15123.
- Maltepe, E.; Schmidt, J.V.; Baunoch, D.; Bradfield, C.A.; Simon, M.C. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature 1997, 386, 403–407.
- Han, Y.; Yang, K.; Proweller, A.; Zhou, G.; Jain, M.K.; Ramirez-Bergeron, D.L. Inhibition of ARNT severely compromises endothelial cell viability and function in response to moderate hypoxia. Angiogenesis 2012, 15, 409–420.
- Dunwoodie, S.L. The role of hypoxia in development of the Mammalian embryo. Dev. Cell 2009, 17, 755–773.
- Duan, L.J.; Zhang-Benoit, Y.; Fong, G.H. Endothelium-intrinsic requirement for Hif-2α during vascular development. Circulation 2005, 111, 2227–2232.
- Patterson, A.J.; Zhang, L. Hypoxia and fetal heart development. Curr. Mol. Med. 2010, 10, 653–666.
- Aitola, M.H.; Pelto-Huikko, M.T. Expression of Arnt and Arnt2 mRNA in developing murine tissues. J. Histochem. Cytochem. 2003, 51, 41–54.
- Xu, B.; Doughman, Y.; Turakhia, M.; Jiang, W.; Landsettle, C.E.; Agani, F.H.; Semenza, G.L.; Watanabe, M.; Yang, Y.C. Partial rescue of defects in Cited2-deficient embryos by HIF-1α heterozygosity. Dev. Biol. 2007, 301, 130–140.
- Armstrong, E.J.; Bischoff, J. Heart valve development: Endothelial cell signaling and differentiation. Circ. Res. 2004, 95, 459–470.
- Tomanek, R.J. Formation of the coronary vasculature during development. Angiogenesis 2005, 8, 273–284.
- Licht, A.H.; Muller-Holtkamp, F.; Flamme, I.; Breier, G. Inhibition of hypoxia-inducible factor activity in endothelial cells disrupts embryonic cardiovascular development. Blood 2006, 107, 584–590.
- Kapitsinou, P.P.; Rajendran, G.; Astleford, L.; Michael, M.; Schonfeld, M.P.; Fields, T.; Shay, S.; French, J.L.; West, J.; Haase, V.H. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell Biol. 2016, 36, 1584–1594.
- Peng, J.; Zhang, L.; Drysdale, L.; Fong, G.H. The transcription factor EPAS-1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 8386–8391.
- Bishop, T.; Ratcliffe, P.J. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ. Res. 2015, 117, 65–79.
- Jiang, X.; Tian, W.; Tu, A.B.; Pasupneti, S.; Shuffle, E.; Dahms, P.; Zhang, P.; Cai, H.; Dinh, T.T.; Liu, B.; et al. Endothelial Hypoxia-Inducible Factor-2α Is Required for the Maintenance of Airway Microvasculature. Circulation 2019, 139, 502–517.
- Abe, H.; Semba, H.; Takeda, N. The Roles of Hypoxia Signaling in the Pathogenesis of Cardiovascular Diseases. J. Atheroscler. Thromb. 2017, 24, 884–894.
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Invest. 2012, 122, 1427–1443.
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial deletion of hypoxia-inducible factor-2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood 2009, 114, 469–477.
- Chen, L.; Endler, A.; Uchida, K.; Horiguchi, S.; Morizane, Y.; Iijima, O.; Toi, M.; Shibasaki, F.; Aarup, A.; Pedersen, T.X.; et al. Int6/eIF3e silencing promotes functional blood vessel outgrowth and enhances wound healing by upregulating hypoxia-induced factor 2α expression. Circulation 2010, 122, 910–919.
- Zhou, Y.; Zhu, X.; Cui, H.; Shi, J.; Yuan, G.; Shi, S.; Hu, Y. The Role of the VEGF Family in Coronary Heart Disease. Front. Cardiovasc. Med. 2021, 8, 738325.
- Ullah, K.; Ai, L.; Li, Y.; Liu, L.; Zhang, Q.; Pan, K.; Humayun, Z.; Sitikov, A.; Su, Q.; Zhao, Q.; et al. A Novel HIF-2α/ARNT Signaling Pathway Protects Against Microvascular Dysfunction and heart failure After Myocardial Infarction. bioRxiv 2023.
- Wu, R.; Chang, H.C.; Khechaduri, A.; Chawla, K.; Tran, M.; Chai, X.; Wagg, C.; Ghanefar, M.; Jiang, X.; Bayeva, M.; et al. Cardiac-specific ablation of ARNT leads to lipotoxicity and cardiomyopathy. J. Clin. Invest. 2014, 124, 4795–4806.
- Knapp, M.; Zheng, M.; Sladojevic, N.; Zhao, Q.; Liao, J.K.; Wu, R. Abstract 20699: Reduction of Endothelial Arnt Mediates Vascular Dysfunction in Diabetes. Circulation 2016, 134, A20699.
- Nyamsuren, G.; Rapp, G.; Dihazi, H.; Zeisberg, E.M.; Tampe, D.; Tampe, B.; Zeisberg, M. PP2A phosphatase inhibition is anti-fibrotic through Ser77 phosphorylation-mediated ARNT/ARNT homodimer formation. Sci. Rep. 2021, 11, 24075.
- Scott, C.; Stokes, R.; Cha, K.M.; Clouston, A.; Eslam, M.; Metwally, M.; Swarbrick, M.M.; George, J.; Gunton, J.E. Myeloid cell deletion of Aryl hydrocarbon Receptor Nuclear Translocator (ARNT) induces non-alcoholic steatohepatitis. PLoS ONE 2019, 14, e0225332.
- Allison, S.J. Targeting ARNT to attenuate renal fibrosis. Nat. Rev. Nephrol. 2018, 14, 535.
- Tampe, B.; Tampe, D.; Nyamsuren, G.; Klöpper, F.; Rapp, G.; Kauffels, A.; Lorf, T.; Zeisberg, E.M.; Müller, G.A.; Kalluri, R.; et al. Pharmacological induction of hypoxia-inducible transcription factor ARNT attenuates chronic kidney failure. J. Clin. Investig. 2018, 128, 3053–3070.
- Su, E.J.; Xin, H.; Yin, P.; Dyson, M.; Coon, J.; Farrow, K.N.; Mestan, K.K.; Ernst, L.M. Impaired Fetoplacental Angiogenesis in Growth-Restricted Fetuses With Abnormal Umbilical Artery Doppler Velocimetry Is Mediated by Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT). J. Clin. Endocrinol. Metab. 2015, 100, E30–E40.
- Bernhardt, W.M.; Campean, V.; Kany, S.; Jurgensen, J.S.; Weidemann, A.; Warnecke, C.; Arend, M.; Klaus, S.; Gunzler, V.; Amann, K.; et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J. Am. Soc. Nephrol. 2006, 17, 1970–1978.
- Hill, P.; Shukla, D.; Tran, M.G.; Aragones, J.; Cook, H.T.; Carmeliet, P.; Maxwell, P.H. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. 2008, 19, 39–46.
- Eckle, T.; Kohler, D.; Lehmann, R.; El Kasmi, K.; Eltzschig, H.K. Hypoxia-inducible factor-1 is central to cardioprotection: A new paradigm for ischemic preconditioning. Circulation 2008, 118, 166–175.
- Bautista, L.; Castro, M.J.; Lopez-Barneo, J.; Castellano, A. Hypoxia inducible factor-2α stabilization and maxi-K+ channel beta1-subunit gene repression by hypoxia in cardiac myocytes: Role in preconditioning. Circ. Res. 2009, 104, 1364–1372.
- Hyvarinen, J.; Hassinen, I.E.; Sormunen, R.; Maki, J.M.; Kivirikko, K.I.; Koivunen, P.; Myllyharju, J. Hearts of hypoxia-inducible factor prolyl 4-hydroxylase-2 hypomorphic mice show protection against acute ischemia-reperfusion injury. J. Biol. Chem. 2010, 285, 13646–13657.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
2 times
(View History)
Update Date:
26 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No