Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Minhyeok Lee | -- | 4427 | 2023-07-25 13:33:43 | | | |
| 2 | Lindsay Dong | Meta information modification | 4427 | 2023-07-26 05:45:42 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Choi, S.R.; Lee, M. Transformer Architecture and Attention Mechanisms in Genome Data. Encyclopedia. Available online: https://encyclopedia.pub/entry/47250 (accessed on 06 August 2026).
Choi SR, Lee M. Transformer Architecture and Attention Mechanisms in Genome Data. Encyclopedia. Available at: https://encyclopedia.pub/entry/47250. Accessed August 06, 2026.
Choi, Sanghyuk Roy, Minhyeok Lee. "Transformer Architecture and Attention Mechanisms in Genome Data" Encyclopedia, https://encyclopedia.pub/entry/47250 (accessed August 06, 2026).
Choi, S.R., & Lee, M. (2023, July 25). Transformer Architecture and Attention Mechanisms in Genome Data. In Encyclopedia. https://encyclopedia.pub/entry/47250
Choi, Sanghyuk Roy and Minhyeok Lee. "Transformer Architecture and Attention Mechanisms in Genome Data." Encyclopedia. Web. 25 July, 2023.
Copy Citation
The emergence and rapid development of deep learning, specifically transformer-based architectures and attention mechanisms, have had transformative implications across several domains, including bioinformatics and genome data analysis. The analogous nature of genome sequences to language texts has enabled the application of techniques that have exhibited success in fields ranging from natural language processing to genomic data.
deep learning
transformer model
attention mechanism
genome data
1. Introduction
The revolution of deep learning methodologies has invigorated the field of bioinformatics and genome data analysis, establishing a foundation for ground-breaking advancements and novel insights [1][2][3][4][5][6]. Recently, the development and application of transformer-based architectures and attention mechanisms have demonstrated superior performance and capabilities in handling the inherent complexity of genome data. Deep learning techniques, particularly those utilizing transformer architectures and attention mechanisms, have shown remarkable success in various domains such as natural language processing (NLP) [7] and computer vision [8][9][10]. These accomplishments have motivated their rapid adoption into bioinformatics, given the similar nature of genome sequences to language texts. Genome sequences can be interpreted as the language of biology, and thus, tools proficient in handling language data can potentially decipher the hidden patterns within these sequences.
The attention mechanism, first introduced in sequence-to-sequence models [11], has revolutionized how deep learning models handle and interpret data [12][13][14][15][16][17][18]. This technique was designed to circumvent the limitations of traditional recurrent models by providing a mechanism to attend to different parts of the input sequence when generating the output. In the context of genome data, this implies the ability to consider different genomic regions and their relations dynamically during the interpretation process. The attention mechanism computes a weighted sum of input features, where the weights, also known as attention scores, are dynamically determined based on the input data. This mechanism allows the model to focus more on essential or relevant features and less on irrelevant or less important ones.
Inspired by the success of attention mechanisms, the transformer model was proposed as a complete shift from the sequential processing nature of recurrent neural networks (RNNs) and their variants [19][20][21][22]. The transformer model leverages attention mechanisms to process the input data in parallel, allowing for faster and more efficient computations. The architecture of the transformer model is composed of a stack of identical transformer modules, each with two sub-layers: a multi-head self-attention mechanism and a position-wise fully connected feed-forward network. Using this architecture, transformer models can capture the dependencies between inputs and outputs without regard for their distance in the sequence.
The potential of transformer-based architectures and attention mechanisms in genome data analysis is vast and largely unexplored. They present a promising solution to tackle the massive scale and intricate nature of genomic data. The ability to capture long-range dependencies between genomic positions, consider multiple relevant genomic regions simultaneously, and adaptively focus on salient features makes these methods uniquely suited for genomic applications.
2. Deep Learning with Transformers and Attention Mechanism
2.1. Conventional Architectures of Deep Learning
In recent years, the field of biomedicine has observed a significant upsurge in the application of machine learning and, more particularly, deep learning methods. These advanced techniques have been instrumental in unearthing insights from complex biomedical datasets, enabling progress in disease diagnosis, drug discovery, and genetic research.
Deep learning, or deep neural networks (DNNs), employs artificial neural networks with multiple layers, a feature that makes it remarkably capable of learning complex patterns from large datasets [23]. One of the simplest forms of a neural network is the multilayer perceptron (MLP), which contains an input layer, one or more hidden layers, and an output layer. MLPs are proficient at handling datasets where inputs and outputs share a linear or non-linear relationship. However, they are less effective when dealing with spatial or temporal data, a limitation overcome by more sophisticated deep learning models such as convolutional neural networks (CNNs) [24] and RNNs [25].
CNNs are exceptionally efficient at processing spatial data, such as images, due to their ability to capture local dependencies in data using convolutional layers. In biomedicine, CNNs have proved instrumental in tasks like medical image analysis and tissue phenotyping.
RNNs, including their advanced variant, long short-term memory (LSTM) networks, are designed to handle sequential data by incorporating a memory-like mechanism, allowing them to learn from previous inputs in the sequence. This property makes them valuable in predicting protein sequences or understanding genetic sequences in bioinformatics.
Generative adversarial networks (GANs), a game-changer in the field, consist of two neural networks, the generator and the discriminator, that compete [26][27][28][29][30][31][32]. This unique architecture enables the generation of new, synthetic data instances that resemble the training data, a feature that holds promise in drug discovery and personalized medicine.
Several other variants of deep learning techniques also exist. For instance, graph attention leverages the attention mechanism to weigh the influence of nodes in a graph, playing a crucial role in molecular biology for structure recognition. Residual networks (ResNets) use shortcut connections to solve the problem of vanishing gradients in deep networks, a feature that can be valuable in medical image analysis. AdaBoost, a boosting algorithm, works by combining multiple weak classifiers to create a strong classifier. Seq2Vec is an approach for sequence data processing where the sequence is converted into a fixed-length vector representation. Finally, variational autoencoders (VAE) are generative models that can learn a latent representation of the input data, offering significant potential in tasks like anomaly detection or dimensionality reduction in complex biomedical data.
2.2. Transformers and Attention Mechanism
The transformer model represents a watershed moment in the evolution of deep learning models [33]. Distinct from conventional sequence transduction models, which typically involve recurrent or convolutional layers, the transformer model solely harnesses attention mechanisms, setting a new precedent in tasks such as machine translation and natural language processing (NLP).
The principal component of a transformer model is the attention mechanism, and it comes in two forms: self-attention (also referred to as intra-attention) and multi-head attention. The attention mechanism’s core function is to model interactions between different elements in a sequence, thereby capturing the dependencies among them without regard to their positions in the sequence. In essence, it determines the extent to which to pay attention to various parts of the input when producing a particular output.
Self-attention mechanisms operate by creating a representation of each element in a sequence that captures the impact of all other elements in the sequence. This is achieved by computing a score for each pair of elements, applying a softmax function to obtain weights, and then using these weights to form a weighted sum of the original element representations. Consequently, it allows each element in the sequence to interact with all other elements, providing a more holistic picture of the entire sequence.
The multi-head attention mechanism, on the other hand, is essentially multiple self-attention mechanisms, or heads, operating in parallel. Each head independently computes a different learned linear transformation of the input, and their outputs are concatenated and linearly transformed to result in the final output. This enables the model to capture various types of relationships and dependencies in the data.
In addition to the self-attention mechanism, another critical aspect of the transformer architecture is the incorporation of positional encoding. Given that the model itself is permutation-invariant (i.e., it does not have any inherent notion of the order of the input elements), there is a necessity for some method to incorporate information about the position of the elements within the sequence. Positional encoding serves this purpose.
Positional encodings are added to the input embeddings at the bottoms of the encoder and decoder stacks. These embeddings are learned or fixed, and their purpose is to inject information about the relative or absolute positions of the words in the sequence. The addition of positional encodings enables the model to make use of the order of the sequence, which is critical for understanding structured data like language.
One common approach to positional encoding is to use sine and cosine functions of different frequencies. With this approach, each dimension of the positional encoding corresponds to a sine or cosine function. These functions have a wavelength that forms a geometric progression from 2𝜋 to 10,000 × 2𝜋.
One of the key advantages of the transformer model is its ability to handle long-range dependencies in the data, an aspect where traditional RNNs and CNNs may struggle due to their sequential nature. By allowing all elements in the sequence to interact simultaneously, transformers alleviate the need for compressing all information into a fixed-size hidden state, which often leads to information loss in long sequences.
Additionally, transformers also introduce the concept of position encoding to counter the absence of inherent positional information in attention mechanisms. This is crucial, especially in tasks where the order of the elements carries significant information.
The transformer’s self-attention mechanism involves three crucial components: the query (Q), key (K), and value (V). These components originate from the input representations and are created by multiplying the input by the respective learned weight matrices. Each of these components carries a unique significance in the attention mechanism.
In detail, the query corresponds to the element for which scholars are trying to compute the context-dependent representation. The key relates to the elements that scholars are comparing the query against to determine the weights. Finally, the value is the element that gets weighted by the attention score (resulting from the comparison of the query with the key) to generate the final output.
The self-attention mechanism operates by calculating an attention score for a pair of query and key. It does so by taking their dot product and then applying a softmax function to ensure that the weights fall into the range of zero and one and sum to one. This provides a normalized measure of importance, or attention, that the model assigns to each element when encoding a particular element.
Following the calculation of attention scores, the model computes a weighted sum of the value vectors, where the weights are given by the attention scores. This operation results in the context-sensitive encoding of each element, where the context depends on all other elements in the sequence. Such encodings are then used as inputs to the next layer in the transformer model.
The use of the Q, K, and V matrices allows the model to learn to focus on different aspects of the input data and enables it to discern which pieces of information are critical when encoding a particular element. As such, the transformer’s attention mechanism brings a significant degree of flexibility and power to the model, allowing it to handle a wide variety of tasks in an efficient and effective manner. The structures of the transformer architecture and the attention mechanism are depicted in Figure 1.
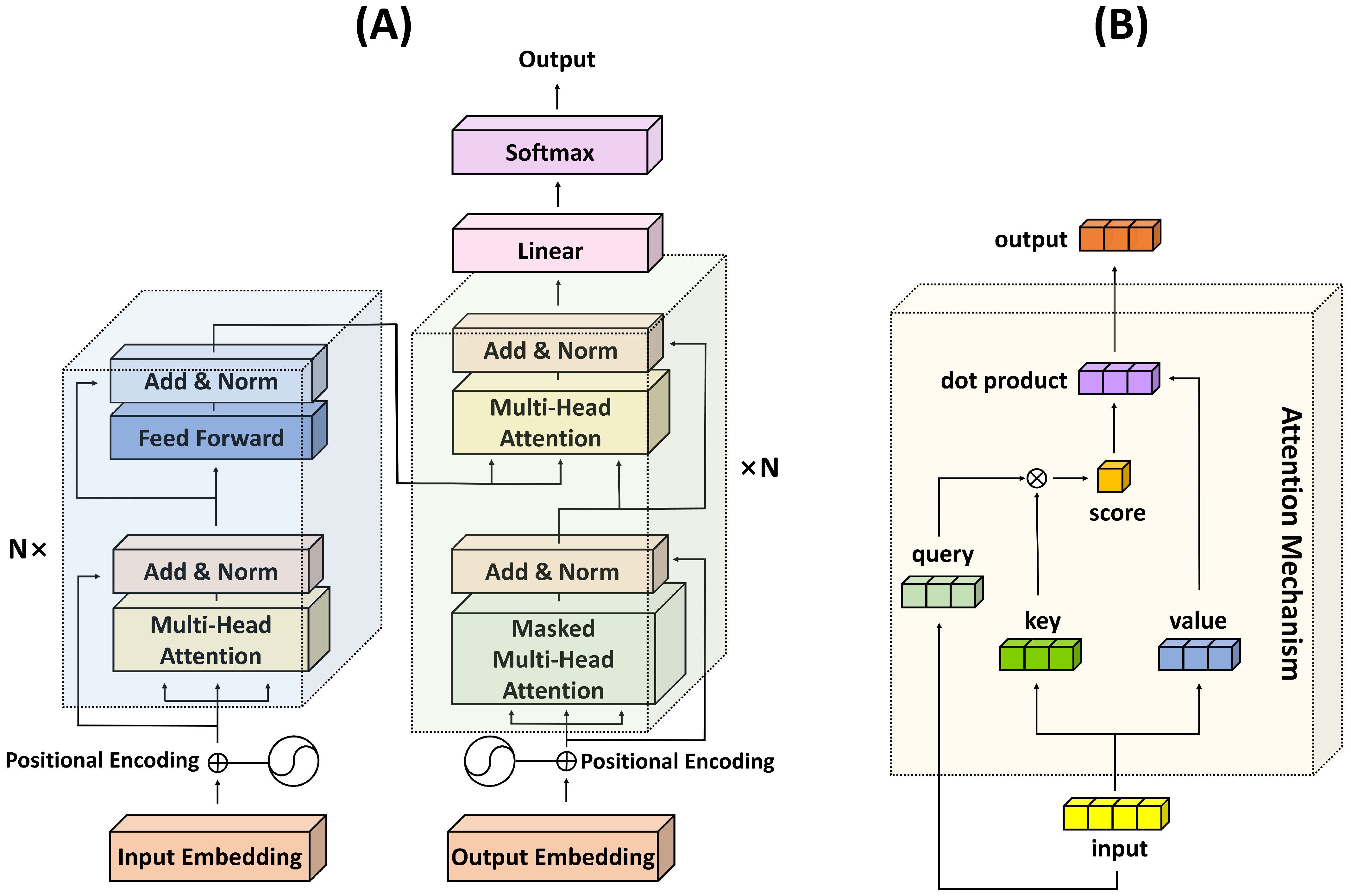
Figure 1. Illustration of the transformer architecture and the attention mechanism. (A) Transformer structure; (B) Attention mechanism.
3. Transformer Architectures and Attention Mechanisms for Genome Data
3.1. Sequence and Site Prediction
In pre-miRNA prediction, Raad et al. [34] introduced miRe2e, a deep learning model based on transformers. The model demonstrated a ten-fold improvement in performance compared to existing algorithms when validated using the human genome. Similarly, Zeng et al. [35] introduced 4mCPred-MTL, a multi-task learning model coupled with a transformer for predicting 4mC sites across multiple species. The model demonstrated a strong feature learning ability, capturing better characteristics of 4mC sites than existing feature descriptors.
Several studies have leveraged deep learning for RNA–protein binding preference prediction. Shen et al. [36] developed a model based on a hierarchical LSTM and attention network which outperformed other methods. Du et al. [37] proposed a deep multi-scale attention network (DeepMSA) based on CNNs to predict the sequence-binding preferences of RNA-binding proteins (RBPs).
An important aspect that distinguishes PrismNet is the application of an attention mechanism that identifies specific RBP-binding nucleotides computationally. The study found enrichment of structure-changing variants (termed riboSNitches) among these dynamic RBP-binding sites, potentially offering new insights into genetic diseases associated with dysregulated RBP bindings. Thus, PrismNet provides a method to access previously inaccessible layers of cell-type-specific RBP–RNA interactions, potentially contributing to our understanding and treatment of human diseases.
In predicting RNA solvent accessibility, Huang et al. [38] proposed a sequence-based model using only primary sequence data. The model employed modified attention layers with different receptive fields to conform to the stem-loop structure of RNA chains. Fan et al. [39] proposed a novel computational method called M(2)pred for accurately predicting the solvent accessibility of RNA. The model utilized a multi-shot neural network with a multi-scale context feature extraction strategy.
To predict transcription factor binding sites, Bhukya et al. [40] proposed two models, PCLAtt and TranAtt. The model outperformed other state-of-the-art methods like DeepSEA, DanQ, TBiNet, and DeepATT in the prediction of binding sites between transcription factors and DNA sequences. Cao et al. [41] proposed DeepARC, an attention-based hybrid approach that combines a CNN and an RNN for predicting transcription factor binding sites.
3.2. Gene Expression and Phenotype Prediction
Deep learning models have been extensively employed to predict gene expression and phenotypes, demonstrating significant improvements over traditional methods. These models have been particularly effective in capturing complex gene–gene and gene–environment interactions and integrating diverse types of genomic and epigenomic data.
A particularly noteworthy study in gene expression and phenotype prediction is that of Angenent-Mari et al. [42]. Their work explores the application of DNNs for the prediction of the function of toehold switches, which serve as a vital model in synthetic biology. These switches, engineered RNA elements, can detect small molecules, proteins, and nucleic acids. However, the prediction of their behavior has posed a considerable challenge—a situation that Angenent-Mari and colleagues sought to address through enhanced pattern recognition from deep learning.
The methodology employed by the authors involved the synthesis and characterization of a dataset comprising 91,534 toehold switches, spanning 23 viral genomes and 906 human transcription factors. The DNNs trained on these nucleotide sequences notably outperformed prior state-of-the-art thermodynamic and kinetic models in the prediction of the toehold switch function. Further, the authors introduced human-understandable attention-visualizations (VIS4Map) which facilitated the identification of successful and failure modes. The network architecture comprised MLP, CNN, and LSTM networks trained on various inputs, including one-hot encoded sequences and rational features. An ensemble MLP model was also proposed, incorporating both the one-hot encoded sequences and rational features.
The advantages of this method are manifold. The authors leveraged deep learning to predict the function of toehold switches, a task that had previously presented considerable challenges. The outperformance of prior state-of-the-art models is a testament to the efficacy of the proposed approach. Furthermore, the inclusion of VIS4Map attention-visualizations enhances the interpretability of the model, providing valuable insights into the model’s workings and facilitating the identification of areas of success and those that need improvement. Despite these significant strides, the methodology also bears certain limitations. The training process is computationally demanding, necessitating high-capacity hardware and graphic processing units which may not be accessible to all researchers.
A key area of focus has been the prediction of gene expression based on histone modifications. Lee et al. [43] developed Chromoformer, a transformer-based deep learning architecture considering large genomic windows and three-dimensional chromatin interactions.
3.3. ncRNA and circRNA Studies
The application of deep learning models, particularly those incorporating transformer architectures and attention mechanisms, has been extensively explored in the study of non-coding RNAs (ncRNAs) and circular RNAs (circRNAs). These models have shown promising results in predicting ncRNA-disease associations, lncRNA–protein interactions, and circRNA-RBP interactions, among other tasks.
Yang et al. [44] presented a novel computational method called iCircRBP-DHN that leverages a deep hierarchical network to distinguish circRNA–RBP-binding sites. The core of this approach is a combination of a deep multi-scale residual network and bidirectional gated recurrent units (BiGRUs) equipped with a self-attention mechanism. This architecture simultaneously extracts local and global contextual information from circRNA sequences. The study proposed two novel encoding schemes to enrich the feature representations. The first, KNFP (K-tuple Nucleotide Frequency Pattern), is designed to capture local contextual features at various scales, effectively addressing the information insufficiency issue inherent in conventional one-hot representation. The second, CircRNA2Vec, is based on the Doc2Vec algorithm and aims to capture global contextual features by modeling long-range dependencies in circRNA sequences. This method treats sequences as a language and maps subsequences (words) into distributed vectors, which contribute to capturing the semantics and syntax of these sequences. The effectiveness of iCircRBP-DHN was validated on multiple circRNAs and linear RNAs datasets, and it showed superior performance over state-of-the-art algorithms.
While iCircRBP-DHN exhibits several advantages, it also presents potential limitations. The method’s strengths include its ability to model both local and global contexts within sequences, its robustness against numerical instability, and its scalability, demonstrated by the performance on extensive datasets. However, the method’s performance is heavily reliant on the quality of sequence data and the effectiveness of the CircRNA2Vec and KNFP encoding schemes, which might not capture all nuances of circRNA–RBP interactions. While the self-attention mechanism can provide some insights into what the model deems important, it might not provide a full explanation of the reasoning behind the model’s predictions.
3.4. Transcription Process Insights
In recent advancements, deep learning, specifically attention mechanisms and transformer models, have been significantly employed in decoding the transcription process of genome data. Clauwaert et al. [45], Park et al. [46], and Han et al. [47] have proposed transformative models centered on transcription factor (TF)-binding site prediction and characterization.
As one of the specific examples, Yan et al. [48] introduced an innovative deep learning framework for circRNA–RBP-binding site discrimination, referred to as iCircRBP-DHN, Integrative Circular RNA–RBP-binding sites Discrimination by Hierarchical Networks. They addressed common issues with previous computational models, such as poor scalability and numerical instability, and developed a transformative method that amalgamates local and global contextual information via deep multi-scale residual network BiGRUs with a self-attention mechanism.
One of the key advantages of this approach is the fusion of two encoding schemes, CircRNA2Vec and the K-tuple nucleotide frequency pattern, which allows for the representation of different degrees of nucleotide dependencies, enhancing the discriminative power of feature representations. The robustness and superior performance of this method were evidenced through extensive testing on 37 circRNA datasets and 31 linear RNA datasets, where it outperformed other state-of-the-art algorithms.
Another interesting application of deep learning is seen in the study by Feng et al. [49], where they developed a model, PEPMAN, that predicts RNA polymerase II pausing sites based on NET-seq data, which are data from a high-throughput technique used to precisely map and quantify nascent transcriptional activity across the genome. PEPMAN utilized attention mechanisms to decipher critical sequence features underlying the pausing of Pol II. Their model’s predictions, in association with various epigenetic features, delivered enlightening insights into the transcription elongation process.
3.5. Multi-Omics/Modal Tasks
Exploring and integrating multi-omics and multi-modal data are substantial tasks in understanding complex biological systems. Deep learning methods, particularly attention mechanisms and transformer models, have seen profound advancements and deployments in this regard. Studies by Gong et al. [50], Kayikci and Khoshgoftaar [51], Ye et al. [52], and Wang et al. [53] have extensively utilized such methods for biomedical data classification and disease prediction.
In the study by Kang et al. [54], a comprehensive ensemble deep learning model for plant miRNA–lncRNA interaction prediction is proposed, namely PmliPEMG. This method introduces a fusion of complex features, multi-scale convolutional long short-term memory (ConvLSTM) networks, and attention mechanisms. Complex features, built using non-linear transformations of sequence and structure features, enhance the sample information at the feature level. By forming a matrix from the complex feature vector, the ConvLSTM models are used as the base model, which is beneficial due to their ability to extract and memorize features over time. Notably, the models are trained on three matrices with different scales, thus enhancing sample information at the scale level.
An attention mechanism layer is incorporated into each base model, assigning different weights to the output of the LSTM layer. This attentional layer allows the model to focus on crucial information during training. Finally, an ensemble method based on a greedy fuzzy decision strategy is implemented to integrate the three base models, improving efficiency and generalization ability. This approach exhibits considerable advantages.
3.6. CRISPR Efficacy and Outcome Prediction
The efficacy and outcome prediction of CRISPR-Cas9 gene editing have significantly improved due to the development of sophisticated deep learning models. Several studies, including Liu et al. [55], Wan and Jiang [56], Xiao et al. [57], Mathis et al. [58], Zhang et al. [59], and Zhang et al. [60], have extensively used such models to predict CRISPR-Cas9 editing outcomes, single guide RNAs (sgRNAs) knockout efficacy, and off-target activities, enhancing the precision of gene editing technologies.
The research by Zhang et al. [60] introduced a novel method for predicting on-target and off-target activities of CRISPR/Cas9 sgRNAs. They proposed two deep learning models, CRISPR-ONT and CRISPR-OFFT, which incorporate an attention-based CNN to focus on sequence elements most decisive in sgRNA efficacy. These models offer several key advantages. First, they utilize an embedding layer that applies k-mer encoding to transform sgRNA sequences into numerical values, allowing the CNN to extract feature maps. This technique has been demonstrated to outperform other methods in sequential analysis. Second, these models use attention mechanisms to improve both prediction power and interpretability, focusing on the elements of the input sequence that are the most relevant to the output. This mirrors how RNA-guide Cas9 nucleases scan the genome, enhancing the realism of the model.
3.7. Gene Regulatory Network Inference
The emergence of deep learning has revolutionized the inference of gene regulatory networks (GRNs) from single-cell RNA-sequencing (scRNA-seq) data, underscoring the utility of transformative machine learning architectures such as the attention mechanism and transformers. Prominent studies, including Lin and Ou-Yang [61], Xu et al. [62], Feng et al. [63], Ullah and Ben-Hur [64], and Xie et al. [65], have utilized these architectures to devise models for GRN inference, highlighting their superior performance compared to conventional methodologies.
The study by Ullah and Ben-Hur [64] presented a novel model, SATORI, for the inference of GRNs. SATORI is a Self-ATtentiOn-based model engineered to detect regulatory element interactions. SATORI leverages the power of deep learning through an amalgamation of convolutional layers and a self-attention mechanism. The convolutional layers, assisted by activation and max-pooling, process the input genomic sequences represented through one-hot encoding. The model further incorporates an optional RNN layer with long short-term memory units for temporal information capture across the sequence.
The multi-head self-attention layer in SATORI is its most pivotal component, designed to model dependencies within the input sequence irrespective of their relative distances. This feature enables the model to effectively capture transcription factor cooperativity. The model is trained and evaluated through a random search algorithm for hyperparameter tuning and the area under the ROC curve for performance measurement. One of the most distinctive features of SATORI is its ability to identify interactions between sequence motifs, contributing to its interpretability. It uses integrated gradients to calculate attribution scores for motifs in a sequence. Changes in these scores after motif mutation can suggest potential interactions.
3.8. Disease Prognosis Estimation
Deep learning models with transformer architectures and attention mechanisms have seen significant utilization in estimating disease prognosis, demonstrating their efficacy in extracting meaningful patterns from complex genomic data. Among the trailblazing studies in this area include those conducted by Lee [66], Choi and Lee [67], Dutta et al. [68], Xing et al. [69], and Meng et al. [70].
Lee [66] introduced the Gene Attention Ensemble NETwork (GAENET), a model designed for prognosis estimation of low-grade glioma (LGG). GAENET incorporated a gene attention mechanism tailored for gene expression data, outperforming traditional methods and identifying HILS1 as the most significant prognostic gene for LGG. Similarly, Choi and Lee [67] proposed Multi-PEN, a deep learning model that utilizes multi-omics and multi-modal schemes for LGG prognosis. The model incorporated gene attention layers for each data type, such as mRNA and miRNA, to identify prognostic genes, showing robust performance compared to existing models.
3.9. Gene Expression-Based Classification
The implementation of deep learning models with transformer architectures and attention mechanisms has significantly improved the classification accuracy based on gene expressions, as presented in numerous studies by Gokhale et al. [71], Beykikhoshk et al. [72], Manica et al. [73], and Lee et al. [74].
Gokhale et al. [71] put forth GeneViT, a vision transformer method, which is a deep learning architecture that applies the principles of self-attention and transformer models to visual data for classifying cancerous gene expressions. This innovative approach started with a dimensionality reduction step using a stacked autoencoder, followed by an improved DeepInsight algorithm, which is a method to transform non-image data to be used for convolution neural network architectures, achieving a remarkable performance edge over existing methodologies, as observed from evaluations on ten benchmark datasets.
3.10. Proteomics
The utilization of deep learning, particularly the incorporation of transformer architectures and attention mechanisms in proteomics, has led to groundbreaking developments in the prediction of protein functionality, as depicted in the studies by Hou et al. [75], Gong et al. [76], Armenteros et al. [77], and Littmann et al. [78].
Hou et al. [75] constructed iDeepSubMito, a deep neural network model designed for the prediction of protein submitochondrial localization. This model employed an inventive graph embedding layer that assimilated interactome data as prior information for prediction. Additionally, an attention layer was incorporated for the integration of various omics features while considering their interactions. The effectiveness of this model was validated by its outperformance of other computational methods during cross-validation on two datasets containing proteins from four mitochondrial compartments.
3.11. Cell-Type Identification
In recent studies, the application of transformer architectures and attention mechanisms in deep learning has brought significant progress to cell-type identification, demonstrating superior performance across various cell types, species, and sequencing depths. The application of transformer architectures and attention mechanisms in deep learning for cell-type identification has seen significant advancements, as evidenced in the studies by Song et al. [79], Feng et al. [80], Buterez et al. [81], and Zhang et al. [82].
Song et al. [79] developed TransCluster, a hybrid network structure that leverages linear discriminant analysis and a modified transformer for enhancing feature learning in single-cell transcriptomic maps. This method outperformed known techniques on various cell datasets from different human tissues, demonstrating high accuracy and robustness.
3.12. Predicting Drug–Drug Interactions
Recent studies have showcased the remarkable progress in predicting drug–drug interactions (DDIs) through the use of deep learning models incorporating transformer architecturesmand attention mechanisms, surpassing classical and other deep learning methods while highlighting significant drug substructures. Deep learning with transformer architectures and attention mechanisms has significantly advanced the prediction of DDIs. Schwarz et al. [83] introduced AttentionDDI, a Siamese self-attention multi-modal neural network that integrates various drug similarity measures derived from drug characteristics. It demonstrated competitive performance compared to state-of-the-art DDI models on multiple benchmark datasets. Similarly, Kim et al. [84] developed DeSIDE-DDI, a framework that incorporates drug-induced gene expression signatures for DDI prediction. This model excelled with an AUC of 0.889 and an Area Under the Precision–Recall (AUPR) of 0.915, surpassing other leading methods in unseen interaction prediction.
References
- Auslander, N.; Gussow, A.B.; Koonin, E.V. Incorporating Machine Learning into Established Bioinformatics Frameworks. Int. J. Mol. Sci. 2021, 22, 2903.
- Lee, M. Deep Learning Techniques with Genomic Data in Cancer Prognosis: A Comprehensive Review of the 2021–2023 Literature. Biology 2023, 12, 893.
- Gomes, R.; Paul, N.; He, N.; Huber, A.F.; Jansen, R.J. Application of Feature Selection and Deep Learning for Cancer Prediction Using DNA Methylation Markers. Genes 2022, 13, 1557.
- Sadad, T.; Aurangzeb, R.A.; Safran, M.; Imran; Alfarhood, S.; Kim, J. Classification of Highly Divergent Viruses from DNA/RNA Sequence Using Transformer-Based Models. Biomedicines 2023, 11, 1323.
- Lee, M. Recent Advances in Deep Learning for Protein-Protein Interaction Analysis: A Comprehensive Review. Molecules 2023, 28, 5169.
- Kim, Y.; Lee, M. Deep Learning Approaches for lncRNA-Mediated Mechanisms: A Comprehensive Review of Recent Developments. Int. J. Mol. Sci. 2023, 24, 10299.
- Brown, T.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.D.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 2020, 33, 1877–1901.
- Khan, S.; Naseer, M.; Hayat, M.; Zamir, S.W.; Khan, F.S.; Shah, M. Transformers in vision: A survey. ACM Comput. Surv. (CSUR) 2022, 54, 1–41.
- Liu, Z.; Lin, Y.; Cao, Y.; Hu, H.; Wei, Y.; Zhang, Z.; Lin, S.; Guo, B. Swin transformer: Hierarchical vision transformer using shifted windows. In Proceedings of the IEEE/CVF International Conference on Computer Vision, Montreal, BC, Canada, 11–17 October 2021; pp. 10012–10022.
- Han, K.; Wang, Y.; Chen, H.; Chen, X.; Guo, J.; Liu, Z.; Tang, Y.; Xiao, A.; Xu, C.; Xu, Y.; et al. A survey on vision transformer. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 45, 87–110.
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30, 6000–6010.
- Wei, Z.; Yan, Q.; Lu, X.; Zheng, Y.; Sun, S.; Lin, J. Compression Reconstruction Network with Coordinated Self-Attention and Adaptive Gaussian Filtering Module. Mathematics 2023, 11, 847.
- Jin, A.; Zeng, X. A Novel Deep Learning Method for Underwater Target Recognition Based on Res-Dense Convolutional Neural Network with Attention Mechanism. J. Mar. Sci. Eng. 2023, 11, 69.
- Gao, L.; Wu, Y.; Yang, T.; Zhang, X.; Zeng, Z.; Chan, C.K.D.; Chen, W. Research on Image Classification and Retrieval Using Deep Learning with Attention Mechanism on Diaspora Chinese Architectural Heritage in Jiangmen, China. Buildings 2023, 13, 275.
- Lu, J.; Ren, H.; Shi, M.; Cui, C.; Zhang, S.; Emam, M.; Li, L. A Novel Hybridoma Cell Segmentation Method Based on Multi-Scale Feature Fusion and Dual Attention Network. Electronics 2023, 12, 979.
- Cheng, S.; Liu, Y. Research on Transportation Mode Recognition Based on Multi-Head Attention Temporal Convolutional Network. Sensors 2023, 23, 3585.
- Kasgari, A.B.; Safavi, S.; Nouri, M.; Hou, J.; Sarshar, N.T.; Ranjbarzadeh, R. Point-of-Interest Preference Model Using an Attention Mechanism in a Convolutional Neural Network. Bioengineering 2023, 10, 495.
- Raimundo, A.; Pavia, J.P.; Sebastião, P.; Postolache, O. YOLOX-Ray: An Efficient Attention-Based Single-Staged Object Detector Tailored for Industrial Inspections. Sensors 2023, 23, 4681.
- Kim, T.; Pak, W. Deep Learning-Based Network Intrusion Detection Using Multiple Image Transformers. Appl. Sci. 2023, 13, 2754.
- Feng, S.; Zhu, X.; Ma, S.; Lan, Q. GIT: A Transformer-Based Deep Learning Model for Geoacoustic Inversion. J. Mar. Sci. Eng. 2023, 11, 1108.
- Jiang, D.; Shi, G.; Li, N.; Ma, L.; Li, W.; Shi, J. TRFM-LS: Transformer-Based Deep Learning Method for Vessel Trajectory Prediction. J. Mar. Sci. Eng. 2023, 11, 880.
- Cao, L.; Wang, Q.; Hong, J.; Han, Y.; Zhang, W.; Zhong, X.; Che, Y.; Ma, Y.; Du, K.; Wu, D.; et al. MVI-TR: A Transformer-Based Deep Learning Model with Contrast-Enhanced CT for Preoperative Prediction of Microvascular Invasion in Hepatocellular Carcinoma. Cancers 2023, 15, 1538.
- Shrestha, A.; Mahmood, A. Review of deep learning algorithms and architectures. IEEE Access 2019, 7, 53040–53065.
- Bhatt, D.; Patel, C.; Talsania, H.; Patel, J.; Vaghela, R.; Pandya, S.; Modi, K.; Ghayvat, H. CNN variants for computer vision: History, architecture, application, challenges and future scope. Electronics 2021, 10, 2470.
- Yu, Y.; Si, X.; Hu, C.; Zhang, J. A review of recurrent neural networks: LSTM cells and network architectures. Neural Comput. 2019, 31, 1235–1270.
- Lee, M.; Seok, J. Controllable generative adversarial network. IEEE Access 2019, 7, 28158–28169.
- Kim, J.; Lee, M. Portfolio optimization using predictive auxiliary classifier generative adversarial networks. Eng. Appl. Artif. Intell. 2023, 125, 106739.
- Lee, M.; Seok, J. Score-guided generative adversarial networks. Axioms 2022, 11, 701.
- Lee, M.; Seok, J. Estimation with uncertainty via conditional generative adversarial networks. Sensors 2021, 21, 6194.
- Yeom, T.; Lee, M. DuDGAN: Improving Class-Conditional GANs via Dual-Diffusion. arXiv 2023, arXiv:2305.14849.
- Ko, K.; Lee, M. ZIGNeRF: Zero-shot 3D Scene Representation with Invertible Generative Neural Radiance Fields. arXiv 2023, arXiv:2306.02741.
- Lee, M. Recent Advances in Generative Adversarial Networks for Gene Expression Data: A Comprehensive Review. Mathematics 2023, 11, 3055.
- Niu, Z.; Zhong, G.; Yu, H. A review on the attention mechanism of deep learning. Neurocomputing 2021, 452, 48–62.
- Raad, J.; Bugnon, L.A.; Milone, D.H.; Stegmayer, G. miRe2e: A full end-to-end deep model based on transformers for prediction of pre-miRNAs. Bioinformatics 2022, 38, 1191–1197.
- Zeng, R.; Cheng, S.; Liao, M. 4mCPred-MTL: Accurate Identification of DNA 4mC Sites in Multiple Species Using Multi-Task Deep Learning Based on Multi-Head Attention Mechanism. Front. Cell Dev. Biol. 2021, 9, 664669.
- Shen, Z.; Zhang, Q.; Han, K.; Huang, D.S. A Deep Learning Model for RNA-Protein Binding Preference Prediction Based on Hierarchical LSTM and Attention Network. IEEE-ACM Trans. Comput. Biol. Bioinform. 2022, 19, 753–762.
- Du, B.; Liu, Z.; Luo, F. Deep multi-scale attention network for RNA-binding proteins prediction. Inf. Sci. 2022, 582, 287–301.
- Huang, Y.; Luo, J.; Jing, R.; Li, M. Multi-model predictive analysis of RNA solvent accessibility based on modified residual attention mechanism. Brief. Bioinform. 2022, 23, bbac470.
- Fan, X.Q.; Hu, J.; Tang, Y.X.; Jia, N.X.; Yu, D.J.; Zhang, G.J. Predicting RNA solvent accessibility from multi-scale context feature via multi-shot neural network. Anal. Biochem. 2022, 654, 114802.
- Bhukya, R.; Kumari, A.; Dasari, C.M.; Amilpur, S. An attention-based hybrid deep neural networks for accurate identification of transcription factor binding sites. Neural Comput. Appl. 2022, 34, 19051–19060.
- Cao, L.; Liu, P.; Chen, J.; Deng, L. Prediction of Transcription Factor Binding Sites Using a Combined Deep Learning Approach. Front. Oncol. 2022, 12, 893520.
- Angenent-Mari, N.M.; Garruss, A.S.; Soenksen, L.R.; Church, G.; Collins, J.J. A deep learning approach to programmable RNA switches. Nat. Commun. 2020, 11, 5057.
- Lee, D.; Yang, J.; Kim, S. Learning the histone codes with large genomic windows and three-dimensional chromatin interactions using transformer. Nat. Commun. 2022, 13, 6678.
- Yang, Y.; Hou, Z.; Ma, Z.; Li, X.; Wong, K.C. iCircRBP-DHN: Identification of circRNA-RBP interaction sites using deep hierarchical network. Brief. Bioinform. 2021, 22, bbaa274.
- Clauwaert, J.; Menschaert, G.; Waegeman, W. Explainability in transformer models for functional genomics. Brief. Bioinform. 2021, 22, bbab060.
- Park, S.; Koh, Y.; Jeon, H.; Kim, H.; Yeo, Y.; Kang, J. Enhancing the interpretability of transcription factor binding site prediction using attention mechanism. Sci. Rep. 2020, 10, 13413.
- Han, K.; Shen, L.C.; Zhu, Y.H.; Xu, J.; Song, J.; Yu, D.J. MAResNet: Predicting transcription factor binding sites by combining multi-scale bottom-up and top-down attention and residual network. Brief. Bioinform. 2022, 23, bbab445.
- Yan, Z.; Lecuyer, E.; Blanchette, M. Prediction of mRNA subcellular localization using deep recurrent neural networks. Bioinformatics 2019, 35, I333–I342.
- Feng, P.; Xiao, A.; Fang, M.; Wan, F.; Li, S.; Lang, P.; Zhao, D.; Zeng, J. A machine learning-based framework for modeling transcription elongation. Proc. Natl. Acad. Sci. USA 2021, 118, e2007450118.
- Gong, P.; Cheng, L.; Zhang, Z.; Meng, A.; Li, E.; Chen, J.; Zhang, L. Multi-omics integration method based on attention deep learning network for biomedical data classification. Comput. Methods Prog. Biomed. 2023, 231, 107377.
- Kayikci, S.; Khoshgoftaar, T.M. Breast cancer prediction using gated attentive multimodal deep learning. J. Big Data 2023, 10, 1–11.
- Ye, L.; Zhang, Y.; Yang, X.; Shen, F.; Xu, B. An Ovarian Cancer Susceptible Gene Prediction Method Based on Deep Learning Methods. Front. Cell Dev. Biol. 2021, 9, 730475.
- Wang, C.; Lye, X.; Kaalia, R.; Kumar, P.; Rajapakse, J.C. Deep learning and multi-omics approach to predict drug responses in cancer. BMC Bioinform. 2022, 22, 1–15.
- Kang, Q.; Meng, J.; Shi, W.; Luan, Y. Ensemble Deep Learning Based on Multi-level Information Enhancement and Greedy Fuzzy Decision for Plant miRNA-lncRNA Interaction Prediction. Interdiscip.-Sci.-Comput. Life Sci. 2021, 13, 603–614.
- Liu, X.; Wang, S.; Ai, D. Predicting CRISPR/Cas9 Repair Outcomes by Attention-Based Deep Learning Framework. Cells 2022, 11, 1847.
- Wan, Y.; Jiang, Z. TransCrispr: Transformer Based Hybrid Model for Predicting CRISPR/Cas9 Single Guide RNA Cleavage Efficiency. IEEE-ACM Trans. Comput. Biol. Bioinform. 2023, 20, 1518–1528.
- Xiao, L.M.; Wan, Y.Q.; Jiang, Z.R. AttCRISPR: A spacetime interpretable model for prediction of sgRNA on-target activity. BMC Bioinform. 2021, 22, 1–17.
- Mathis, N.; Allam, A.; Kissling, L.; Marquart, K.F.; Schmidheini, L.; Solari, C.; Balazs, Z.; Krauthammer, M.; Schwank, G. Predicting prime editing efficiency and product purity by deep learning. Nat. Biotechnol. 2023.
- Zhang, Z.R.; Jiang, Z.R. Effective use of sequence information to predict CRISPR-Cas9 off-target. Comput. Struct. Biotechnol. J. 2022, 20, 650–661.
- Zhang, G.; Zeng, T.; Dai, Z.; Dai, X. Prediction of CRISPR/Cas9 single guide RNA cleavage efficiency and specificity by attention-based convolutional neural networks. Comput. Struct. Biotechnol. J. 2021, 19, 1445–1457.
- Lin, Z.; Ou-Yang, L. Inferring gene regulatory networks from single-cell gene expression data via deep multi-view contrastive learning. Brief. Bioinform. 2022, 24, bbac586.
- Xu, J.; Zhang, A.; Liu, F.; Zhang, X. STGRNS: An interpretable transformer-based method for inferring gene regulatory networks from single-cell transcriptomic data. Bioinformatics 2023, 39, btad165.
- Feng, X.; Fang, F.; Long, H.; Zeng, R.; Yao, Y. Single-cell RNA-seq data analysis using graph autoencoders and graph attention networks. Front. Genet. 2022, 13, 1003711.
- Ullah, F.; Ben-Hur, A. A self-attention model for inferring cooperativity between regulatory features. Nucleic Acids Res. 2021, 49, e77.
- Xie, X.; Wang, Y.; Sheng, N.; Zhang, S.; Cao, Y.; Fu, Y. Predicting miRNA-disease associations based on multi-view information fusion. Front. Genet. 2022, 13, 979815.
- Lee, M. An Ensemble Deep Learning Model with a Gene Attention Mechanism for Estimating the Prognosis of Low-Grade Glioma. Biology 2022, 11, 586.
- Choi, S.R.; Lee, M. Estimating the Prognosis of Low-Grade Glioma with Gene Attention Using Multi-Omics and Multi-Modal Schemes. Biology 2022, 11, 1462.
- Dutta, P.; Patra, A.P.; Saha, S. DeePROG: Deep Attention-Based Model for Diseased Gene Prognosis by Fusing Multi-Omics Data. IEEE-ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2770–2781.
- Xing, X.; Yang, F.; Li, H.; Zhang, J.; Zhao, Y.; Gao, M.; Huang, J.; Yao, J. Multi-level attention graph neural network based on co-expression gene modules for disease diagnosis and prognosis. Bioinformatics 2022, 38, 2178–2186.
- Meng, X.; Wang, X.; Zhang, X.; Zhang, C.; Zhang, Z.; Zhang, K.; Wang, S. A Novel Attention-Mechanism Based Cox Survival Model by Exploiting Pan-Cancer Empirical Genomic Information. Cells 2022, 11, 1421.
- Gokhale, M.; Mohanty, S.K.; Ojha, A. GeneViT: Gene Vision Transformer with Improved DeepInsight for cancer classification. Comput. Biol. Med. 2023, 155, 106643.
- Beykikhoshk, A.; Quinn, T.P.; Lee, S.C.; Tran, T.; Venkatesh, S. DeepTRIAGE: Interpretable and individualised biomarker scores using attention mechanism for the classification of breast cancer sub-types. BMC Med. Genom. 2020, 13, 1–10.
- Manica, M.; Oskooei, A.; Born, J.; Subramanian, V.; Saez-Rodriguez, J.; Martinez, M.R. Toward Explainable Anticancer Compound Sensitivity Prediction via Multimodal Attention-Based Convolutional Encoders. Mol. Pharm. 2019, 16, 4797–4806.
- Lee, S.; Lim, S.; Lee, T.; Sung, I.; Kim, S. Cancer subtype classification and modeling by pathway attention and propagation. Bioinformatics 2020, 36, 3818–3824.
- Hou, Z.; Yang, Y.; Li, H.; Wong, K.C.; Li, X. iDeepSubMito: Identification of protein submitochondrial localization with deep learning. Brief. Bioinform. 2021, 22, bbab288.
- Gong, H.; Wen, J.; Luo, R.; Feng, Y.; Guo, J.; Fu, H.; Zhou, X. Integrated mRNA sequence optimization using deep learning. Brief. Bioinform. 2023, 24, bbad001.
- Armenteros, J.J.A.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2019, 2, e201900429.
- Littmann, M.; Heinzinger, M.; Dallago, C.; Weissenow, K.; Rost, B. Protein embeddings and deep learning predict binding residues for various ligand classes. Sci. Rep. 2021, 11, 23916.
- Song, T.; Dai, H.; Wang, S.; Wang, G.; Zhang, X.; Zhang, Y.; Jiao, L. TransCluster: A Cell-Type Identification Method for single-cell RNA-Seq data using deep learning based on transformer. Front. Genet. 2022, 13, 1038919.
- Feng, X.; Zhang, H.; Lin, H.; Long, H. Single-cell RNA-seq data analysis based on directed graph neural network. Methods 2023, 211, 48–60.
- Buterez, D.; Bica, I.; Tariq, I.; Andres-Terre, H.; Lio, P. CellVGAE: An unsupervised scRNA-seq analysis workflow with graph attention networks. Bioinformatics 2022, 38, 1277–1286.
- Zhang, Y.; Blanchette, M. Reference panel guided topological structure annotation of Hi-C data. Nat. Commun. 2022, 13, 7426.
- Schwarz, K.; Allam, A.; Gonzalez, N.A.P.; Krauthammer, M. AttentionDDI: Siamese attention-based deep learning method for drug-drug interaction predictions. BMC Bioinform. 2021, 22, 1–19.
- Kim, E.; Nam, H. DeSIDE-DDI: Interpretable prediction of drug-drug interactions using drug-induced gene expressions. J. Cheminform. 2022, 14, 1–12.
More
Information
Subjects:
Mathematical & Computational Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.1K
Revisions:
2 times
(View History)
Update Date:
26 Jul 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No