 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antonio Paoli | -- | 3239 | 2023-07-25 10:41:09 | | | |
| 2 | Lindsay Dong | Meta information modification | 3239 | 2023-07-26 02:46:00 | | |
Video Upload Options
Together with the global rise in obesity and metabolic syndrome, the prevalence of individuals who suffer from nonalcoholic fatty liver disease (NAFLD) has risen dramatically. NAFLD is currently the most common chronic liver disease and includes a continuum of liver disorders from initial fat accumulation to nonalcoholic steatohepatitis (NASH), considered the more severe forms, which can evolve in, cirrhosis, and hepatocellular carcinoma. Common features of NAFLD includes altered lipid metabolism mainly linked to mitochondrial dysfunction, which, as a vicious cycle, aggravates oxidative stress and promotes inflammation and, as a consequence, the progressive death of hepatocytes and the severe form of NAFLD. A ketogenic diet (KD), i.e., a diet very low in carbohydrates (<30 g/die) that induces “physiological ketosis”, has been demonstrated to alleviate oxidative stress and restore mitochondrial function.
1. Introduction
2. NAFLD
3. Ketogenic Diet
4. KD and Mitochondria
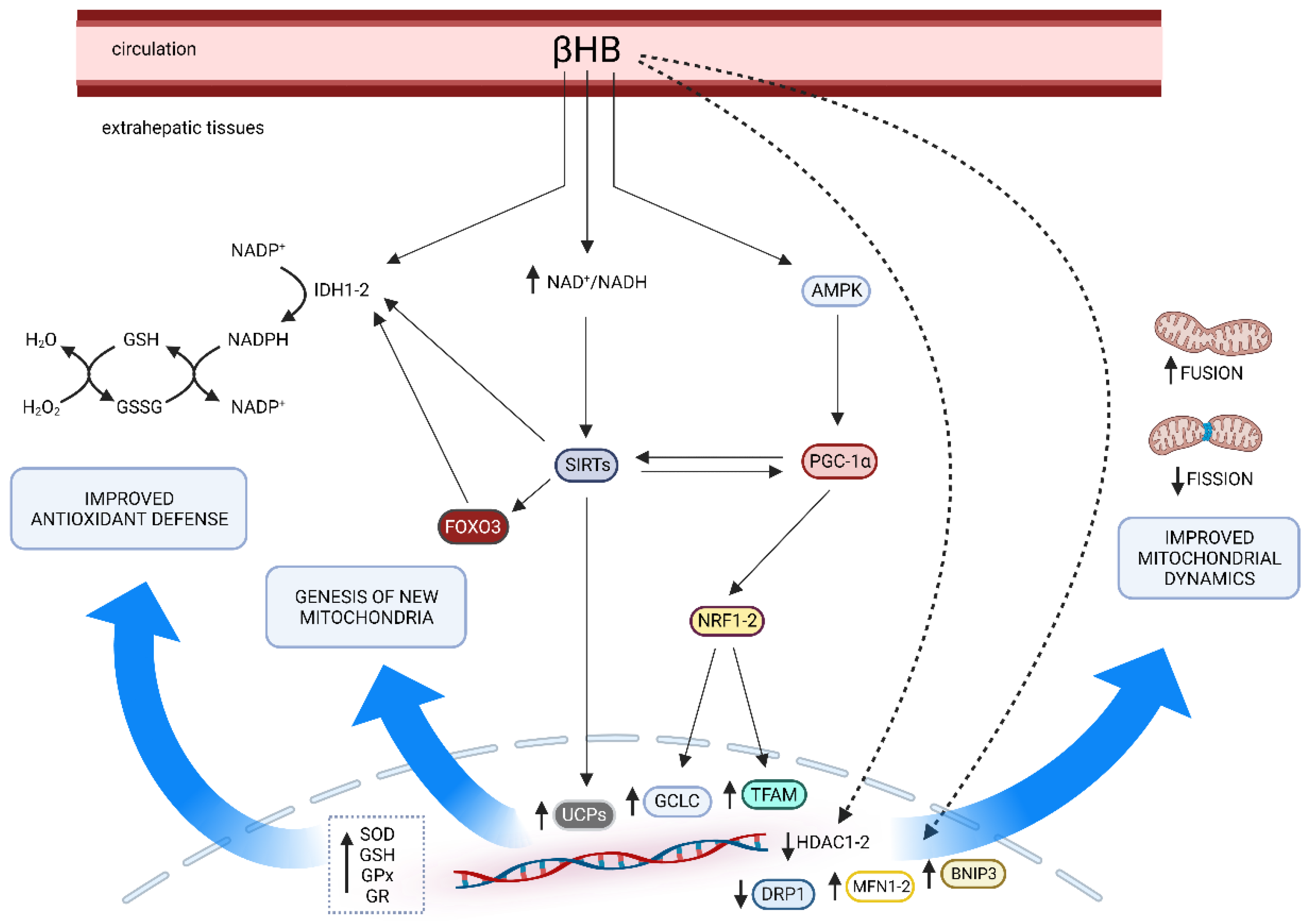
5. Effects of Ketosis on Oxidative Stress Pathways
6. Liver and Mitochondria
7. Liver and Oxidative Stress
8. KD and Liver
References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13.
- EASL–EASD–EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402.
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209.
- Li, Y.; Xie, Z.; Song, Q.; Li, J. Mitochondria Homeostasis: Biology and Involvement in Hepatic Steatosis to NASH. Acta Pharm. Sin. 2022, 43, 1141–1155.
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Free Radic. Biol. Med. 2020, 152, 116–141.
- Gonzalez, A.; Huerta-Salgado, C.; Orozco-Aguilar, J.; Aguirre, F.; Tacchi, F.; Simon, F.; Cabello-Verrugio, C. Role of Oxidative Stress in Hepatic and Extrahepatic Dysfunctions during Nonalcoholic Fatty Liver Disease (NAFLD). Oxid. Med. Cell. Longev. 2020, 2020, 1617805.
- Longo, M.; Meroni, M.; Paolini, E.; Macchi, C.; Dongiovanni, P. Mitochondrial Dynamics and Nonalcoholic Fatty Liver Disease (NAFLD): New Perspectives for a Fairy-Tale Ending? Metabolism 2021, 117, 154708.
- Cichoz-Lach, H.; Michalak, A. Oxidative Stress as a Crucial Factor in Liver Diseases. World J. Gastroenterol. 2014, 20, 8082–8091.
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613.
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease. Hepatology 2018, 67, 123–133.
- Sanyal, A.J. Past, Present and Future Perspectives in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386.
- Siddiqui, M.S.; Harrison, S.A.; Abdelmalek, M.F.; Anstee, Q.M.; Bedossa, P.; Castera, L.; Dimick-Santos, L.; Friedman, S.L.; Greene, K.; Kleiner, D.E.; et al. Case Definitions for Inclusion and Analysis of Endpoints in Clinical Trials for Nonalcoholic Steatohepatitis through the Lens of Regulatory Science. Hepatology 2018, 67, 2001–2012.
- Bence, K.K.; Birnbaum, M.J. Metabolic Drivers of Non-Alcoholic Fatty Liver Disease. Mol. Metab. 2021, 50, 101143.
- Ziolkowska, S.; Binienda, A.; Jabłkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 11128.
- Phinney, S.D.; Bistrian, B.R.; Evans, W.J.; Gervino, E.; Blackburn, G.L. The Human Metabolic Response to Chronic Ketosis without Caloric Restriction: Preservation of Submaximal Exercise Capability with Reduced Carbohydrate Oxidation. Metabolism 1983, 32, 769–776.
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond Weight Loss: A Review of the Therapeutic Uses of Very-Low-Carbohydrate (Ketogenic) Diets. Eur. J. Clin. Nutr. 2013, 67, 789–796.
- Ashtary-Larky, D.; Bagheri, R.; Bavi, H.; Baker, J.S.; Moro, T.; Mancin, L.; Paoli, A. Ketogenic Diets, Physical Activity and Body Composition: A Review. Br. J. Nutr. 2022, 127, 1898–1920.
- Paoli, A. Ketogenic Diet for Obesity: Friend or Foe? Int. J. Environ. Res. Public Health 2014, 11, 2092–2107.
- Goldenberg, J.Z.; Johnston, B.C. Low and Very Low Carbohydrate Diets for Diabetes Remission. BMJ 2021, 373, n262.
- Harvey, C.J.D.C.; Schofield, G.M.; Zinn, C.; Thornley, S.J.; Crofts, C.; Merien, F.L.R. Low-Carbohydrate Diets Differing in Carbohydrate Restriction Improve Cardiometabolic and Anthropometric Markers in Healthy Adults: A Randomised Clinical Trial. PeerJ 2019, 7, e6273.
- Ilyas, Z.; Perna, S.; Alalwan, T.A.; Zahid, M.N.; Spadaccini, D.; Gasparri, C.; Peroni, G.; Faragli, A.; Alogna, A.; la Porta, E.; et al. The Ketogenic Diet: Is It an Answer for Sarcopenic Obesity? Nutrients 2022, 14, 620.
- Robinson, P.J.; Rapoport, S.I. Glucose Transport and Metabolism in the Brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1986, 250, R127–R136.
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F. Brain Metabolism during Fasting. J. Clin. Investig. 1967, 46, 1589–1595.
- Mitchell, R.W.; Hatch, G.M. Fatty Acid Transport into the Brain: Of Fatty Acid Fables and Lipid Tails. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 293–302.
- Paoli, A.; Cenci, L.; Grimaldi, K.A. Effect of Ketogenic Mediterranean Diet with Phytoextracts and Low Carbohydrates/High-Protein Meals on Weight, Cardiovascular Risk Factors, Body Composition and Diet Compliance in Italian Council Employees. Nutr. J. 2011, 10, 112.
- Krebs, H.A. The Regulation of the Release of Ketone Bodies by the Liver. Adv. Enzym. Regul. 1966, 4, 339–353.
- Paoli, A.; Bianco, A.; Grimaldi, K.A. The Ketogenic Diet and Sport. Exerc. Sport Sci. Rev. 2015, 43, 153–162.
- Veech, R.L. The Therapeutic Implications of Ketone Bodies: The Effects of Ketone Bodies in Pathological Conditions: Ketosis, Ketogenic Diet, Redox States, Insulin Resistance, and Mitochondrial Metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 309–319.
- Pathak, S.J.; Baar, K. Ketogenic Diets and Mitochondrial Function: Benefits for Aging But Not for Athletes. Exerc. Sport Sci. Rev. 2023, 51, 27–33.
- Akbari, M.; Kirkwood, T.B.L.; Bohr, V.A. Mitochondria in the Signaling Pathways That Control Longevity and Health Span. Ageing Res. Rev. 2019, 54, 100940.
- Negro, M.; Cerullo, G.; Parimbelli, M.; Ravazzani, A.; Feletti, F.; Berardinelli, A.; Cena, H.; D’Antona, G. Exercise, Nutrition, and Supplements in the Muscle Carnitine Palmitoyl-Transferase II Deficiency: New Theoretical Bases for Potential Applications. Front. Physiol. 2021, 12, 704290.
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial Dynamics, Mitophagy and Cardiovascular Disease. J. Physiol. 2016, 594, 509–525.
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159.
- Forbes, J.M.; Thorburn, D.R. Mitochondrial Dysfunction in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 291–312.
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharm. Toxicol. 2018, 58, 353–389.
- Rai, S.N.; Singh, C.; Singh, A.; Singh, M.P.; Singh, B.K. Mitochondrial Dysfunction: A Potential Therapeutic Target to Treat Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 3075–3088.
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.v.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Cardiovascular Disease: A Brief Review. Ann. Med. 2018, 50, 121–127.
- Halling, J.F.; Pilegaard, H. PGC-1α-Mediated Regulation of Mitochondrial Function and Physiological Implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936.
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661.
- Imai, S.; Guarente, L. NAD+ and Sirtuins in Aging and Disease. Trends Cell Biol. 2014, 24, 464–471.
- Kyriazis, I.; Vassi, E.; Alvanou, M.; Angelakis, C.; Skaperda, Z.; Tekos, F.; Garikipati, V.; Spandidos, D.; Kouretas, D. The Impact of Diet upon Mitochondrial Physiology (Review). Int. J. Mol. Med. 2022, 50, 135.
- Vidali, S.; Aminzadeh, S.; Lambert, B.; Rutherford, T.; Sperl, W.; Kofler, B.; Feichtinger, R.G. Mitochondria: The Ketogenic Diet—A Metabolism-Based Therapy. Int. J. Biochem. Cell Biol. 2015, 63, 55–59.
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone Bodies: From Enemy to Friend and Guardian Angel. BMC Med. 2021, 19, 313.
- Miller, V.J.; LaFountain, R.A.; Barnhart, E.; Sapper, T.S.; Short, J.; Arnold, W.D.; Hyde, P.N.; Crabtree, C.D.; Kackley, M.L.; Kraemer, W.J.; et al. A Ketogenic Diet Combined with Exercise Alters Mitochondrial Function in Human Skeletal Muscle While Improving Metabolic Health. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E995–E1007.
- Qu, C.; Keijer, J.; Adjobo-Hermans, M.J.W.; van de Wal, M.; Schirris, T.; van Karnebeek, C.; Pan, Y.; Koopman, W.J.H. The Ketogenic Diet as a Therapeutic Intervention Strategy in Mitochondrial Disease. Int. J. Biochem. Cell Biol. 2021, 138, 106050.
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional Ketosis and Mitohormesis: Potential Implications for Mitochondrial Function and Human Health. J. Nutr. Metab. 2018, 2018, 5157645.
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res. 2019, 44, 22–37.
- Abduraman, M.A.; Azizan, N.A.; Teoh, S.H.; Tan, M.L. Ketogenesis and SIRT1 as a Tool in Managing Obesity. Obes. Res. Clin. Pract. 2021, 15, 10–18.
- Tozzi, R.; Cipriani, F.; Masi, D.; Basciani, S.; Watanabe, M.; Lubrano, C.; Gnessi, L.; Mariani, S. Ketone Bodies and SIRT1, Synergic Epigenetic Regulators for Metabolic Health: A Narrative Review. Nutrients 2022, 14, 3145.
- Wallace, M.A.; Aguirre, N.W.; Marcotte, G.R.; Marshall, A.G.; Baehr, L.M.; Hughes, D.C.; Hamilton, K.L.; Roberts, M.N.; Lopez-Dominguez, J.A.; Miller, B.F.; et al. The Ketogenic Diet Preserves Skeletal Muscle with Aging in Mice. Aging Cell 2021, 20, e13322.
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360.
- Sidarala, V.; Zhu, J.; Levi-D’Ancona, E.; Pearson, G.L.; Reck, E.C.; Walker, E.M.; Kaufman, B.A.; Soleimanpour, S.A. Mitofusin 1 and 2 Regulation of Mitochondrial DNA Content Is a Critical Determinant of Glucose Homeostasis. Nat. Commun. 2022, 13, 2340.
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506.
- Newell, C.; Shutt, T.E.; Ahn, Y.; Hittel, D.S.; Khan, A.; Rho, J.M.; Shearer, J. Tissue Specific Impacts of a Ketogenic Diet on Mitochondrial Dynamics in the BTBRT+tf/j Mouse. Front. Physiol. 2016, 7, 654.
- Haces, M.L.; Hernández-Fonseca, K.; Medina-Campos, O.N.; Montiel, T.; Pedraza-Chaverri, J.; Massieu, L. Antioxidant Capacity Contributes to Protection of Ketone Bodies against Oxidative Damage Induced during Hypoglycemic Conditions. Exp. Neurol. 2008, 211, 85–96.
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, Ketone Bodies, and Mitochondrial Energy Transduction. FASEB J. 1995, 9, 651–658.
- Veech, R.L. The Determination of the Redox States and Phosphorylation Potential in Living Tissues and Their Relationship to Metabolic Control of Disease Phenotypes. Biochem. Mol. Biol. Educ. 2006, 34, 168–179.
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a Dietary Ketone Ester on Hippocampal Glycolytic and Tricarboxylic Acid Cycle Intermediates and Amino Acids in a 3xTgAD Mouse Model of Alzheimer’s Disease. J. Neurochem. 2017, 141, 195–207.
- Xin, L.; Ipek, Ö.; Beaumont, M.; Shevlyakova, M.; Christinat, N.; Masoodi, M.; Greenberg, N.; Gruetter, R.; Cuenoud, B. Nutritional Ketosis Increases NAD+/NADH Ratio in Healthy Human Brain: An in Vivo Study by 31P-MRS. Front. Nutr. 2018, 5, 62.
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 Mediates Neuroprotection of Ketones against Ischemic Stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1783–1789.
- Yin, J.; Nielsen, M.; Li, S.; Shi, J. Ketones Improves Apolipoprotein E4-Related Memory Deficiency via Sirtuin 3. Aging 2019, 11, 4579–4586.
- Kashiwaya, Y.; King, M.T.; Veech, R.L. Substrate Signaling by Insulin. Am. J. Cardiol. 1997, 80, 50A–64A.
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones Inhibit Mitochondrial Production of Reactive Oxygen Species Production Following Glutamate Excitotoxicity by Increasing NADH Oxidation. Neuroscience 2007, 145, 256–264.
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate Regulates Energy Metabolism and Induces BDNF Expression in Cerebral Cortical Neurons. J. Neurochem. 2016, 139, 769–781.
- Pasyukova, E.G.; Vaiserman, A.M. HDAC Inhibitors: A New Promising Drug Class in Anti-Aging Research. Mech. Ageing Dev. 2017, 166, 6–15.
- Wang, X.; Wu, X.; Liu, Q.; Kong, G.; Zhou, J.; Jiang, J.; Wu, X.; Huang, Z.; Su, W.; Zhu, Q. Ketogenic Metabolism Inhibits Histone Deacetylase (HDAC) and Reduces Oxidative Stress After Spinal Cord Injury in Rats. Neuroscience 2017, 366, 36–43.
- Li, B.; Yu, Y.; Liu, K.; Zhang, Y.; Geng, Q.; Zhang, F.; Li, Y.; Qi, J. β-Hydroxybutyrate Inhibits Histone Deacetylase 3 to Promote Claudin-5 Generation and Attenuate Cardiac Microvascular Hyperpermeability in Diabetes. Diabetologia 2021, 64, 226–239.
- Guerrieri, F.; Nicoletti, C.; Adorisio, E.; Caraccio, G.; Leonetti, P.; Zanotti, F.; Cantatore, P. Correlation between Decreased Expression of Mitochondrial F0F1-ATP Synthase and Low Regenerating Capability of the Liver after Partial Hepatectomy in Hypothyroid Rats. J. Bioenerg. Biomembr. 2000, 32, 183–191.
- Mashek, D.G. Hepatic Fatty Acid Trafficking: Multiple Forks in the Road. Adv. Nutr. 2013, 4, 697–710.
- Barrows, B.R.; Parks, E.J. Contributions of Different Fatty Acid Sources to Very Low-Density Lipoprotein-Triacylglycerol in the Fasted and Fed States. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452.
- Passarella, S.; de Bari, L.; Valenti, D.; Pizzuto, R.; Paventi, G.; Atlante, A. Mitochondria and L-Lactate Metabolism. FEBS Lett. 2008, 582, 3569–3576.
- Paventi, G.; Pizzuto, R.; Passarella, S. The Occurrence of L-Lactate Dehydrogenase in the Inner Mitochondrial Compartment of Pig Liver. Biochem. Biophys. Res. Commun. 2017, 489, 255–261.
- Fromenty, B.; Roden, M. Mitochondrial Alterations in Fatty Liver Diseases. J. Hepatol. 2023, 78, 415–429.
- Zhang, C.; Zhao, Y.; Yu, M.; Qin, J.; Ye, B.; Wang, Q. Mitochondrial Dysfunction and Chronic Liver Disease. Curr. Issues Mol. Biol. 2022, 44, 3156–3165.
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375.
- Grattagliano, I.; de Bari, O.; Bernardo, T.C.; Oliveira, P.J.; Wang, D.Q.-H.; Portincasa, P. Role of Mitochondria in Nonalcoholic Fatty Liver Disease-from Origin to Propagation. Clin. Biochem. 2012, 45, 610–618.
- Muriel, P.; Gordillo, K.R. Role of Oxidative Stress in Liver Health and Disease. Oxid. Med. Cell. Longev. 2016, 2016, 9037051.
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, Oxidative Stress and Cell Death. Apoptosis 2007, 12, 913–922.
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in Non-Alcoholic Fatty Liver Disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99.
- Zuo, J.; Zhang, Z.; Luo, M.; Zhou, L.; Nice, E.C.; Zhang, W.; Wang, C.; Huang, C. Redox Signaling at the Crossroads of Human Health and Disease. MedComm 2022, 3, e127.
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124.
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728.
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69.
- Figueira, T.R.; Barros, M.H.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.B.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.C.; Vercesi, A.E. Mitochondria as a Source of Reactive Oxygen and Nitrogen Species: From Molecular Mechanisms to Human Health. Antioxid. Redox Signal. 2013, 18, 2029–2074.
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52.
- Zhang, Y.; Wong, H.S. Are Mitochondria the Main Contributor of Reactive Oxygen Species in Cells? J. Exp. Biol. 2021, 224, jeb221606.
- del Río, L.A.; López-Huertas, E. ROS Generation in Peroxisomes and Its Role in Cell Signaling. Plant Cell Physiol. 2016, 57, pcw076.
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462.
- Zheng, W.; Sun, Q.; Li, L.; Cheng, Y.; Chen, Y.; Lv, M.; Xiang, X. Role of Endoplasmic Reticulum Stress in Hepatic Glucose and Lipid Metabolism and Therapeutic Strategies for Metabolic Liver Disease. Int. Immunopharmacol. 2022, 113, 109458.
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540.
- Matuz-Mares, D.; Vázquez-Meza, H.; Vilchis-Landeros, M.M. NOX as a Therapeutic Target in Liver Disease. Antioxidants 2022, 11, 2038.
- de Medeiros, I.C.; de Lima, J.G. Is Nonalcoholic Fatty Liver Disease an Endogenous Alcoholic Fatty Liver Disease?—A Mechanistic Hypothesis. Med. Hypotheses 2015, 85, 148–152.
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity during Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10.
- Parra-Vargas, M.; Rodriguez-Echevarria, R.; Jimenez-Chillaron, J.C. Nutritional Approaches for the Management of Nonalcoholic Fatty Liver Disease: An Evidence-Based Review. Nutrients 2020, 12, 3860.
- Mooli, R.G.R.; Ramakrishnan, S.K. Emerging Role of Hepatic Ketogenesis in Fatty Liver Disease. Front. Physiol. 2022, 13, 946474.
- Sripongpun, P.; Churuangsuk, C.; Bunchorntavakul, C. Current Evidence Concerning Effects of Ketogenic Diet and Intermittent Fasting in Patients with Nonalcoholic Fatty Liver. J. Clin. Transl. Hepatol. 2022, 10, 730–739.
- Watanabe, M.; Tozzi, R.; Risi, R.; Tuccinardi, D.; Mariani, S.; Basciani, S.; Spera, G.; Lubrano, C.; Gnessi, L. Beneficial Effects of the Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Comprehensive Review of the Literature. Obes. Rev. 2020, 21, e13024.
- Tendler, D.; Lin, S.; Yancy, W.S.; Mavropoulos, J.; Sylvestre, P.; Rockey, D.C.; Westman, E.C. The Effect of a Low-Carbohydrate, Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Pilot Study. Dig. Dis. Sci. 2007, 52, 589–593.
- Paoli, A.; Tinsley, G.; Bianco, A.; Moro, T. The Influence of Meal Frequency and Timing on Health in Humans: The Role of Fasting. Nutrients 2019, 11, 719.

