Together with the global rise in obesity and metabolic syndrome, the prevalence of individuals who suffer from nonalcoholic fatty liver disease (NAFLD) has risen dramatically. NAFLD is currently the most common chronic liver disease and includes a continuum of liver disorders from initial fat accumulation to nonalcoholic steatohepatitis (NASH), considered the more severe forms, which can evolve in, cirrhosis, and hepatocellular carcinoma. Common features of NAFLD includes altered lipid metabolism mainly linked to mitochondrial dysfunction, which, as a vicious cycle, aggravates oxidative stress and promotes inflammation and, as a consequence, the progressive death of hepatocytes and the severe form of NAFLD. A ketogenic diet (KD), i.e., a diet very low in carbohydrates (<30 g/die) that induces “physiological ketosis”, has been demonstrated to alleviate oxidative stress and restore mitochondrial function.
- ketogenic diet
- nonalcoholic fatty liver disease (NAFLD)
- mitochondria
- oxidative stress
- liver
- ketone bodies
1. Introduction
2. NAFLD
In the last decade, there has been a growing interest in investigating NAFLD due to its fast global spread. Today, around one in four adults suffer from this condition and it is estimated that the number of people suffering from NAFLD in the United States will double by 2030 [21][11]. This rising rate will be followed by increasing numbers of patients with hepatocellular carcinoma, cirrhosis and liver failure. In general, the term NAFLD refers to a wide variety of liver disorders, from simple steatosis, where fat liver infiltration is still relatively low, to nonalcoholic steatohepatitis (NASH), considered the more severe end of the disease spectrum, where liver function (i.e., lipid metabolism) and anatomy (i.e., fibrosis) may be severely compromised [22][12]. Currently, liver biopsy remains the gold standard method for the definitive diagnosis of NAFL or NASH [23][13]. In NASH, hepatic steatosis is accompanied by lobular inflammation and exacerbated hepatocyte damage, promoting fibrosis and cirrhosis. Based on level of severity, it’s possible to distinguish from mild fibrosis (stage 1) to bridging fibrosis (stage 4) [1]. The NAFLD’s pathogenesis is still not completely understood but the main commonly accepted theories include adipose liver infiltration, abnormalities of hepatocyte metabolism, mitochondria disfunction, altered hepatic immune cell function, and systemic inflammation. According to the more simplistic “two hit” hypothesis, the “first hit” derive from insulin resistance and altered lipid metabolism (increased hepatic lipogenesis and impaired FFA degradation), which causes liver steatosis. Thus, this condition sensitizes the liver to further metabolic insults (“second hit”) that led to OxS, activation of inflammation processes and fibrogenesis resulting in the progression of liver disease. However, hepatic steatosis is often associated with overweight/obesity (especially the excess of visceral adiposity) or metabolic dysregulation such as type 2 diabetes mellitus, elevated triacylglycerols, decreased high-density lipoprotein cholesterol or increased blood pressure [27,28][14][15]. In this scenario, the term MAFLD has recently been suggested to be more appropriate to describe the metabolic dysfunction associated with liver disease [4].3. Ketogenic Diet
The ketogenic diet (KD) is a nutritional pattern characterized by a high content of fat and adequate protein content but a very low carbohydrate intake (less than 20 g d−1 or 5% of total daily energy intake) [29][16]. This macronutrient distribution forces the body to use fat as its primary fuel source, resulting in physiological ketosis (i.e., blood ketone bodies concentrations higher than 0.3 or 0.5 mmol/L and blood pH within the physiological range as a consequence of the increase of ketone bodies (KB) production [30,31][17][18]. Growing evidence suggests that carbohydrate-restricted diets, such as KD, can be properly used in several condition from health to disease, i.e., obesity, diabetes, dyslipidemia, hypertension, neurological disorders, and many cancers [30,32,33,34,35][17][19][20][21][22]. Glucose is widely considered the main energy provider for brain’s metabolism [36][23]. However, after a period of few days of fasting or KD, glucose storages (glycogen in muscles and liver) become insufficient to support the energy needs of the central nervous system and to sustain fat oxidation process [37][24]. Indeed, oxaloacetate (an unstable, fundamental intermediate of the Krebs cycle) cannot be accumulated and stored, but must be produced primarily (in mammalians) from the conversion of glucose to pyruvate and, then to oxaloacetate through a, so called, anaplerotic reaction. Despite it is generally accepted that fatty acids cannot cross the blood-brain barrier, some data suggest instead that a certain amount of fatty acids can pass the blood-brain barrier even in a limited quantity [38][25]. It is critical to emphasize that, despite the deprivation of carbohydrates, blood glucose levels remain physiologically stable due to gluconeogenesis [42][26], which involves glucogenic amino acids as well (especially in individuals with obesity) as glycerol derived from TGs. In healthy people, this state, where KB levels can rise up to 7 to 8 mmol L−1 without any pH change, is considered a physiological adaptation [43][27]. Instead in the case of pathological diabetic ketoacidosis, extremely high ketonemia (>20 mmol L−1), lowering of blood pH (<7.3), and high blood glycemia coexist [44][28].4. KD and Mitochondria
The effect of KBs on mitochondrial function is thought to be among the major contributors to the benefits induced by the KD and the underlying mechanisms are currently under investigation [40,45][29][30]. Mitochondria are the most studied organelles in the energy-production system of a cell [46[31][32],47], essentially known for ATP generation through the Krebs cycle in the mitochondrial matrix and the oxidative phosphorylation (OXPHOS) in the mitochondrial inner membrane. In addition, mitochondria are even implicated in other vital cell activities such as redox balance, calcium homeostasis, and apoptosis regulation [46,48,49][31][33][34]. It is not unexpected that variations in mitochondrial activity have been associated to a wide range of diseases such as metabolic and degenerative disorders, epilepsy, and cancer [48,50,51,52,53][33][35][36][37][38]. Abnormal mitochondrial functions generally include an impaired OXPHOS, defective mitochondrial dynamics and altered mitochondrial biogenesis strictly linked to a decline in the activity of different regulators of mitochondrial health such as sirtuins (SIRT1-7) or peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α) [51][36]. PGC-1α is commonly considered as a master regulator of biogenesis as well as mitochondrial quality control mechanisms, i.e., mitochondrial remodeling (fission and fusion cycle) and autophagy (mitophagy) [54][39]; SIRTs are class III nicotinamide-adenine-dinucleotide (NAD+)-dependent histone deacetylases (HDACs) implicated in many signaling that modulate metabolic pathways, redox homeostasis, proliferation, and maintenance of genome stability [55,56][40][41]. Mitochondria are dynamic structures and their number and function are regulated by the interaction between internal factors and external variables through complex mechanisms. Among external variables, food and physical exercise are the most important factors able to influence mitochondria physiology [57][42]. As underlined by many authors, KD can promote mitochondrial health by improving mitochondrial activity, stimulating the genesis of new mitochondria and remodeling [15,45,57,58,59,60,61][30][42][43][44][45][46][47]. The KD forces the body to use fat as its main source of fuel, as mentioned above. Consequently, the KD inevitably stimulates numerous pathways, upregulating key proteins involved in the OXPHOS system as well as the Krebs cycle (citrate synthase and malate dehydrogenase), FA oxidation (carnitine palmitoyl-transferase, long and very-long chain acyl-coA dehydrogenase and β-hydroxyacyl-coA dehydrogenase) leading to an increase of all these bioenergetic process and mitochondrial activity in general [61][47]. In agreement with these results, Hasan-Olive and colleagues has recently proposed that mitochondrial dysfunction could be reversed via the PGC1α-SIRT3-UCP2 axis activation in mice on a KD [65][48]. During KD or fasting, KB and SIRT1 converge on PGC-1α, acting as direct molecular drivers of epigenomic reprogramming through histone modifications and promoting gluconeogenesis, higher FA oxidation and mitochondrial biogenesis [66,67][49][50]. For instance, Wallace and colleagues showed that 14 months of KD increased the expression of the PGC-1α, SIRT1, SIRT3, and proteins from each complex of the electron transport chain, resulting in increases in mitochondrial biogenesis and antioxidant activity concomitant with a mitigation of age-related muscle loss compared to the control group [68][51]. Lastly, maintaining mitochondrial morphology is fundamental to preserving or restoring proper function, and KD is associated with better mitochondrial dynamics. Briefly, cycles of fission and fusion are orchestrated mainly by three proteins: MFN1 and MFN2 (mitofusin 1-2) are considered essential for mitochondrial fusion, while DRP1 (dynamin-related protein 1) is required for mitochondrial fission [75,76][52][53]. In cases of energy restriction, such as starvation, acute stress, and senescence, increased fusion and/or decreased fission activity promotes mitochondria lengthening [77][54]. On the other hand, enhanced fission with reduced fusion activity results in the shortening of mitochondria and, thus, impaired bioenergetic activity [77][54]. Furthermore, it seems that KD may even act as a mitophagy activator. Selective elimination of damaged mitochondria via mitochondria-specific autophagy (“mitophagy”) is essential for mitochondrial health, and the expression levels of BNIP3, a mitophagy regulator gene, have been reported to be upregulated in mice fed the KD [82][55]. Considering the above, it can be suggested that KD may slow or reduce mitochondrial dysfunctions. The multiple effects of KDs (especially βHB) on mitochondrial functions are summarized in Figure 1.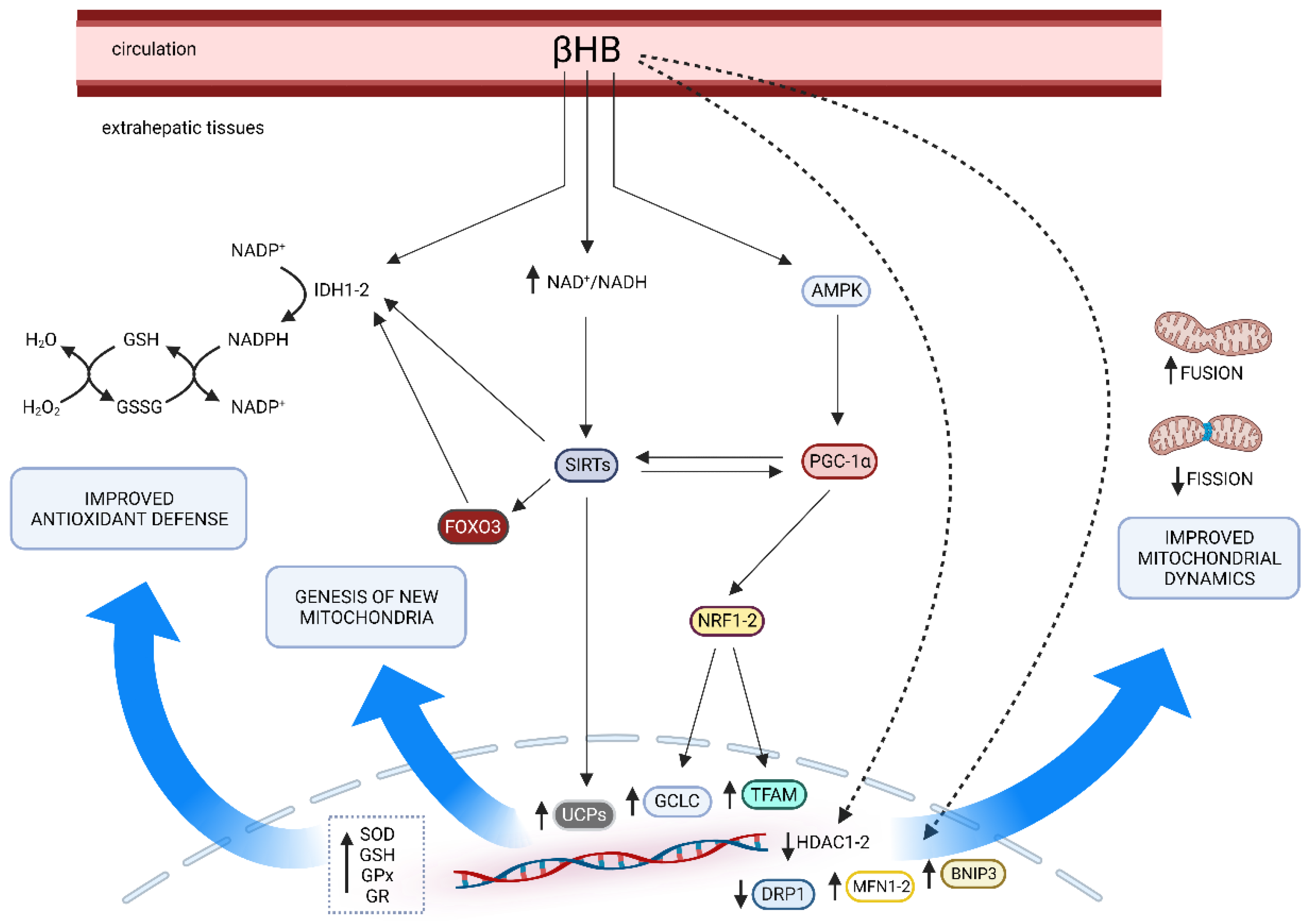
5. Effects of Ketosis on Oxidative Stress Pathways
Although it is generally recognized that ketosis decreases the overall OxS, the exact mechanisms are still poorly understood. KB may directly impact OxS; for example, βHB acts as a scavenger for hydroxyl radicals (•OH) due to the presence of the hydroxyl group in βHB [83][56]. On the other hand, KB could improve the cell’s redox state indirectly in several ways. Among these, studies from Veech and colleagues reported an improvement of mitochondrial function through the increase of the redox span between complex I and II due to decreased reduction of free mitochondrial NAD+, leading to an increased NAD+/NADH ratio and the increase of free mitochondrial CoQ/CoQH ratio [84,85][57][58]. During ketosis, an increase in the estimated energy available in the transport of electrons from mitochondrial NAD to Q couple may minimize H2O2 generation [84][57]. The positive effects of KB on the OxS, via the increase of NAD+/NADH ratio, has been widely documented in animal [86,87,88,89][59][60][61][62] ex vivo [90][63] and cellular models [91,92][64][65]. During ketosis, the enhanced NAD+/NADH ratio is associated with an increased AMP/ATP ratio, due to a rapid ATP depletion, especially under caloric restriction [61][47]. Thus, the activation of AMPK observed under KD could be related to a high AMP/ATP ratio, at least until compensatory events take place (e.g., improved ATP generation). As proposed by Kolb and colleagues, is ketolysis itself to promote a cellular response, via the activation of AMP-activated protein kinase (AMPK), NRF2, and SIRTs, considered key factors of energetic pathways and other cell-protective activities such as anti-oxidant and anti-inflammatory response, as also stated in the other paragraph [58][44] Cellular metabolites may also influence gene expression through their activity as cofactors for epigenetic modifications mediated by histones and the effects of KB on OxS could be partially related to those mechanisms associated to the histone acetylation. Inside the cell, transcriptional activity can be modulated by specific epigenome modifiers called histone deacetylases (HDACs, EC 3.5.1.98). Normally, histone acetyltransferase (EC 2.3.1.48) is modulated by nuclear acetyl-CoA concentration whilst NAD+ concentration regulates SIRTs [98][66]. The higher concentration of Acetil CoA and KB, due to nutritional ketosis of fasting, can influence the acetylation status of histones. Findings from Wang et al. demonstrated a 31–43% inhibition of HDAC activity after 2 weeks of KD in a rat model of spinal cord injury [99][67]. Similar results have been observed after 10 weeks treatment with βHB in a diabetic rat model [100][68].6. Liver and Mitochondria
Mitochondria have a fundamental role in the regulation of hepatic cellular redox metabolism, and its lipid metabolism. In fact, mitochondria occupy about 18% of the hepatocytes volume playing a pivotal role in several signaling pathways linked to fat metabolism [116][69]. The liver is essential for numerous physiological processes. One of these is lipid metabolism: from digestive absorption (via biliary synthesis and secretion) to FFA homeostasis, including process of synthesis, oxidation, and lipid storage (mainly in form of triglycerides, TG). The liver accumulates FFA in the following ways: (1) by uptaking circulating FFA derived from lipolysis of TG in adipocytes (about 60–80%) and from chylomicron remnant (about 15%); (2) via de novo synthesis of FFA (about 5–25%). (1) A large part of the FFA pool in the liver derives from the uptake of FFA obtained from adipocytes’ TG through lipolysis. Lipolysis is the process by which TG are hydrolyzed to FFA and glycerol. This process, at the level of adipocytes, involves the sequential action of several lipases, such as adipose TG lipase, hormone-sensitive lipase, and monoglyceride lipase. Then, due to their hydrophobic nature, FFA are transported mainly bounded to albumin from the adipose tissue (storage site) to the other tissues and organs that utilize it (i.e., the liver) [117][70]. On the other hand, a small portion of the liver FFA pool is formed from dietary FFA after the digestion process in the small intestine where lipids are emulsified by bile salts. TG are then re-synthesized by enterocytes and transported into the lymph (exocytosis) and blood as lipoprotein particles (chylomicrons with cholesteryl esters, phospholipids, and the apolipoprotein ApoB-48). It was reported that the uptake of chylomicron remnants accounts for up to 25% of the liver FA pool during the fed state [118][71]. (2) Hepatic de novo lipogenesis is promoted basically by insulin. In fact, both fat and carbohydrates contribute to the FFA pool in the liver. As in other tissues such as in mammary gland, FFA can also be produced from two carbon units (acetyl-CoA). During the process of fatty acid synthesis through acetyl-CoA, mitochondria have a major role: pyruvate, obtained from glucose during glycolysis, can enter the mitochondrion via the mitochondrial pyruvate carrier and, in the matrix, provides Acetyl-CoA, via the pyruvate dehydrogenase complex, and oxaloacetate via the pyruvate carboxylase [119,120][72][73]. Regarding lipid catabolism, firstly, FFA are converted into fatty acyl-CoA by acyl-CoA synthase in the cytosol of hepatocyte. Then, carnitine palmitoyl-transferase 1 catalyzes the reaction from Acyl-CoA + carnitine to CoA and acylcarnitine which can enter mitochondria via the acylcarnitine/L-carnitine antiporter using the L-carnitine shuttle. A second transferase localized at the matrix side of the inner membrane allows the oxidation of acyl-CoA, via the β-oxidation, to acetyl-CoA, while L-carnitine is released. The relevance of mitochondrial function in the liver is also being explored in many conditions, from health to illness [122,123,124,125][74][75][76][77]. Numerous studies have reported altered mitochondrial activity in conditions characterized by liver damage, mainly linked to OxS, poor bioenergetics, and fat accumulation [9,10,124,126][9][10][76][78]. Overproduction of ROS and lipotoxic lipid accumulation can occur in cases of decreased β-oxidation (e.g., in liver steatosis) or electron transfer chain (ETC) impairments [7]. In more dept, during the early stages of lipid metabolism disturbances, mitochondrial activity boosts, as a compensatory response to minimize the harmful effects of increased lipid accumulation [127][79]. The compromised ETC leads to an overgeneration of ROS, decreased ATP synthesis and the oxidative damage of the internal mitochondrial membrane. This impaired integrity, reduces mitochondrial permeability by opening the mitochondrial permeability transition pore, and allows the release of pro-apoptotic factors to the cytosol (e.g., cytochrome C) [129][80].7. Liver and Oxidative Stress
ROS generation plays critical roles in normal physiological processes, modulating cellular homeostasis from health to disease [137][81]. At the same time, an imbalance between the antioxidant system and massive ROS accumulation produces OxS, which promotes an inflammatory response and triggers apoptosis and fibrosis in hepatic tissue [9[9][82],138], leading to liver injury and functional dysfunction [26,139][83][84]. Generally, mitochondria are recognized as the most important ROS producers and are particularly relevant when considering ROS derived from energetic metabolism [140,141][85][86]. Nevertheless, many other sources of non-mitochondrial ROS, have been identified in more recent studies [142][87]. Among these, peroxisomes that produce H2O2 as a normal sub-product of fatty acid oxidation [47,143][32][88]. OxS is regarded as a crucial factor in the progression of liver disease [9[9][36][82][89],51,138,148], and mitochondrial dysfunction, endoplasmic reticulum (ER) stress, and NOX up-regulation are basically the principal mechanisms linked to the overproduction of ROS and OxS [6]. Firstly, mitochondrial activity is directly linked to the energy balance and normal function of hepatocytes, as previously mentioned. At the same time, OxS promotes ER Stress. The ER is an organelle, abundant in hepatocytes, engaged in multiple functions, including lipid metabolism and calcium homeostasis [149][90]. Moreover, the ER is crucial, especially in the synthesis, folding, and modification of proteins [150][91]. Alterations in ER redox balance contribute to metabolic dysfunctions and an increase of unfolded protein response (UPR). The UPR is generally controlled by three transmembrane stress transducer proteins: activating transcription factor 6 (ATF6), inositol-requiring signaling protein 1 (IRE1), and protein kinase RNA-like ER kinase (PERK). ER stress causes a variety of consequences in hepatocytes through its downstream pathways. Overexpression of NOXs contribute, through the generation of ROS, to oxidative damage and hepatic fibrosis by acting through multiple pathways [154][92]. For example, the upregulation of NOX-1 mediates NAFLD-induced endothelial dysfunction in the liver. The ROS excess produced by NOX-1 may reduce the NO bioavailability and affect liver circulation by impairing the vasodilation response and decreasing its anti-inflammatory, antifibrogenic, and antioxidant properties in the endothelium [155][93]. The expression of NOX4 and related ROS generation was significantly increased during development of steatohepatitis in mice [156][94].8. KD and Liver
In recent times, the KD has emerged as an effective nutritional strategy for the management of NAFLD and other related metabolic diseases, such as obesity-associated type 2 diabetes mellitus, which plays a pivotal role in the pathogenesis and progression of NAFLD [13,18,19,20][95][96][97][98]. For the first time, a pilot study carried out by Tendler et al. demonstrated that six month of a calorie unrestricted KD led to and improvements in steatosis and fibrosis in four obese patients with histological diagnosis of NAFLD together with a significant weight loss (10.9% on average) [167][99]. It is crucial to note, however, that in the majority of these studies, there was no control group and KD was combined with caloric restriction. KD and caloric restriction have many pathways and targets in common, thus, a synergistic effect can’t be excluded [173][100]. Overall, based on available evidence, adjusting the dietary macronutrient composition by simply altering the carbohydrate-fat ratio with or without energy intake limitation is recommended [13][95]. In this regard, KD could be recognized as a promising dietary therapy for NAFLD for several reasons.References
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13.
- EASL–EASD–EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402.
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209.
- Li, Y.; Xie, Z.; Song, Q.; Li, J. Mitochondria Homeostasis: Biology and Involvement in Hepatic Steatosis to NASH. Acta Pharm. Sin. 2022, 43, 1141–1155.
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Free Radic. Biol. Med. 2020, 152, 116–141.
- Gonzalez, A.; Huerta-Salgado, C.; Orozco-Aguilar, J.; Aguirre, F.; Tacchi, F.; Simon, F.; Cabello-Verrugio, C. Role of Oxidative Stress in Hepatic and Extrahepatic Dysfunctions during Nonalcoholic Fatty Liver Disease (NAFLD). Oxid. Med. Cell. Longev. 2020, 2020, 1617805.
- Longo, M.; Meroni, M.; Paolini, E.; Macchi, C.; Dongiovanni, P. Mitochondrial Dynamics and Nonalcoholic Fatty Liver Disease (NAFLD): New Perspectives for a Fairy-Tale Ending? Metabolism 2021, 117, 154708.
- Cichoz-Lach, H.; Michalak, A. Oxidative Stress as a Crucial Factor in Liver Diseases. World J. Gastroenterol. 2014, 20, 8082–8091.
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613.
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease. Hepatology 2018, 67, 123–133.
- Sanyal, A.J. Past, Present and Future Perspectives in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386.
- Siddiqui, M.S.; Harrison, S.A.; Abdelmalek, M.F.; Anstee, Q.M.; Bedossa, P.; Castera, L.; Dimick-Santos, L.; Friedman, S.L.; Greene, K.; Kleiner, D.E.; et al. Case Definitions for Inclusion and Analysis of Endpoints in Clinical Trials for Nonalcoholic Steatohepatitis through the Lens of Regulatory Science. Hepatology 2018, 67, 2001–2012.
- Bence, K.K.; Birnbaum, M.J. Metabolic Drivers of Non-Alcoholic Fatty Liver Disease. Mol. Metab. 2021, 50, 101143.
- Ziolkowska, S.; Binienda, A.; Jabłkowski, M.; Szemraj, J.; Czarny, P. The Interplay between Insulin Resistance, Inflammation, Oxidative Stress, Base Excision Repair and Metabolic Syndrome in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 11128.
- Phinney, S.D.; Bistrian, B.R.; Evans, W.J.; Gervino, E.; Blackburn, G.L. The Human Metabolic Response to Chronic Ketosis without Caloric Restriction: Preservation of Submaximal Exercise Capability with Reduced Carbohydrate Oxidation. Metabolism 1983, 32, 769–776.
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond Weight Loss: A Review of the Therapeutic Uses of Very-Low-Carbohydrate (Ketogenic) Diets. Eur. J. Clin. Nutr. 2013, 67, 789–796.
- Ashtary-Larky, D.; Bagheri, R.; Bavi, H.; Baker, J.S.; Moro, T.; Mancin, L.; Paoli, A. Ketogenic Diets, Physical Activity and Body Composition: A Review. Br. J. Nutr. 2022, 127, 1898–1920.
- Paoli, A. Ketogenic Diet for Obesity: Friend or Foe? Int. J. Environ. Res. Public Health 2014, 11, 2092–2107.
- Goldenberg, J.Z.; Johnston, B.C. Low and Very Low Carbohydrate Diets for Diabetes Remission. BMJ 2021, 373, n262.
- Harvey, C.J.D.C.; Schofield, G.M.; Zinn, C.; Thornley, S.J.; Crofts, C.; Merien, F.L.R. Low-Carbohydrate Diets Differing in Carbohydrate Restriction Improve Cardiometabolic and Anthropometric Markers in Healthy Adults: A Randomised Clinical Trial. PeerJ 2019, 7, e6273.
- Ilyas, Z.; Perna, S.; Alalwan, T.A.; Zahid, M.N.; Spadaccini, D.; Gasparri, C.; Peroni, G.; Faragli, A.; Alogna, A.; la Porta, E.; et al. The Ketogenic Diet: Is It an Answer for Sarcopenic Obesity? Nutrients 2022, 14, 620.
- Robinson, P.J.; Rapoport, S.I. Glucose Transport and Metabolism in the Brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1986, 250, R127–R136.
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F. Brain Metabolism during Fasting. J. Clin. Investig. 1967, 46, 1589–1595.
- Mitchell, R.W.; Hatch, G.M. Fatty Acid Transport into the Brain: Of Fatty Acid Fables and Lipid Tails. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 293–302.
- Paoli, A.; Cenci, L.; Grimaldi, K.A. Effect of Ketogenic Mediterranean Diet with Phytoextracts and Low Carbohydrates/High-Protein Meals on Weight, Cardiovascular Risk Factors, Body Composition and Diet Compliance in Italian Council Employees. Nutr. J. 2011, 10, 112.
- Krebs, H.A. The Regulation of the Release of Ketone Bodies by the Liver. Adv. Enzym. Regul. 1966, 4, 339–353.
- Paoli, A.; Bianco, A.; Grimaldi, K.A. The Ketogenic Diet and Sport. Exerc. Sport Sci. Rev. 2015, 43, 153–162.
- Veech, R.L. The Therapeutic Implications of Ketone Bodies: The Effects of Ketone Bodies in Pathological Conditions: Ketosis, Ketogenic Diet, Redox States, Insulin Resistance, and Mitochondrial Metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 309–319.
- Pathak, S.J.; Baar, K. Ketogenic Diets and Mitochondrial Function: Benefits for Aging But Not for Athletes. Exerc. Sport Sci. Rev. 2023, 51, 27–33.
- Akbari, M.; Kirkwood, T.B.L.; Bohr, V.A. Mitochondria in the Signaling Pathways That Control Longevity and Health Span. Ageing Res. Rev. 2019, 54, 100940.
- Negro, M.; Cerullo, G.; Parimbelli, M.; Ravazzani, A.; Feletti, F.; Berardinelli, A.; Cena, H.; D’Antona, G. Exercise, Nutrition, and Supplements in the Muscle Carnitine Palmitoyl-Transferase II Deficiency: New Theoretical Bases for Potential Applications. Front. Physiol. 2021, 12, 704290.
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial Dynamics, Mitophagy and Cardiovascular Disease. J. Physiol. 2016, 594, 509–525.
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159.
- Forbes, J.M.; Thorburn, D.R. Mitochondrial Dysfunction in Diabetic Kidney Disease. Nat. Rev. Nephrol. 2018, 14, 291–312.
- Sorrentino, V.; Menzies, K.J.; Auwerx, J. Repairing Mitochondrial Dysfunction in Disease. Annu. Rev. Pharm. Toxicol. 2018, 58, 353–389.
- Rai, S.N.; Singh, C.; Singh, A.; Singh, M.P.; Singh, B.K. Mitochondrial Dysfunction: A Potential Therapeutic Target to Treat Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 3075–3088.
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.v.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Cardiovascular Disease: A Brief Review. Ann. Med. 2018, 50, 121–127.
- Halling, J.F.; Pilegaard, H. PGC-1α-Mediated Regulation of Mitochondrial Function and Physiological Implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936.
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661.
- Imai, S.; Guarente, L. NAD+ and Sirtuins in Aging and Disease. Trends Cell Biol. 2014, 24, 464–471.
- Kyriazis, I.; Vassi, E.; Alvanou, M.; Angelakis, C.; Skaperda, Z.; Tekos, F.; Garikipati, V.; Spandidos, D.; Kouretas, D. The Impact of Diet upon Mitochondrial Physiology (Review). Int. J. Mol. Med. 2022, 50, 135.
- Vidali, S.; Aminzadeh, S.; Lambert, B.; Rutherford, T.; Sperl, W.; Kofler, B.; Feichtinger, R.G. Mitochondria: The Ketogenic Diet—A Metabolism-Based Therapy. Int. J. Biochem. Cell Biol. 2015, 63, 55–59.
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone Bodies: From Enemy to Friend and Guardian Angel. BMC Med. 2021, 19, 313.
- Miller, V.J.; LaFountain, R.A.; Barnhart, E.; Sapper, T.S.; Short, J.; Arnold, W.D.; Hyde, P.N.; Crabtree, C.D.; Kackley, M.L.; Kraemer, W.J.; et al. A Ketogenic Diet Combined with Exercise Alters Mitochondrial Function in Human Skeletal Muscle While Improving Metabolic Health. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E995–E1007.
- Qu, C.; Keijer, J.; Adjobo-Hermans, M.J.W.; van de Wal, M.; Schirris, T.; van Karnebeek, C.; Pan, Y.; Koopman, W.J.H. The Ketogenic Diet as a Therapeutic Intervention Strategy in Mitochondrial Disease. Int. J. Biochem. Cell Biol. 2021, 138, 106050.
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional Ketosis and Mitohormesis: Potential Implications for Mitochondrial Function and Human Health. J. Nutr. Metab. 2018, 2018, 5157645.
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res. 2019, 44, 22–37.
- Abduraman, M.A.; Azizan, N.A.; Teoh, S.H.; Tan, M.L. Ketogenesis and SIRT1 as a Tool in Managing Obesity. Obes. Res. Clin. Pract. 2021, 15, 10–18.
- Tozzi, R.; Cipriani, F.; Masi, D.; Basciani, S.; Watanabe, M.; Lubrano, C.; Gnessi, L.; Mariani, S. Ketone Bodies and SIRT1, Synergic Epigenetic Regulators for Metabolic Health: A Narrative Review. Nutrients 2022, 14, 3145.
- Wallace, M.A.; Aguirre, N.W.; Marcotte, G.R.; Marshall, A.G.; Baehr, L.M.; Hughes, D.C.; Hamilton, K.L.; Roberts, M.N.; Lopez-Dominguez, J.A.; Miller, B.F.; et al. The Ketogenic Diet Preserves Skeletal Muscle with Aging in Mice. Aging Cell 2021, 20, e13322.
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial Dynamics: Overview of Molecular Mechanisms. Essays Biochem. 2018, 62, 341–360.
- Sidarala, V.; Zhu, J.; Levi-D’Ancona, E.; Pearson, G.L.; Reck, E.C.; Walker, E.M.; Kaufman, B.A.; Soleimanpour, S.A. Mitofusin 1 and 2 Regulation of Mitochondrial DNA Content Is a Critical Determinant of Glucose Homeostasis. Nat. Commun. 2022, 13, 2340.
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506.
- Newell, C.; Shutt, T.E.; Ahn, Y.; Hittel, D.S.; Khan, A.; Rho, J.M.; Shearer, J. Tissue Specific Impacts of a Ketogenic Diet on Mitochondrial Dynamics in the BTBRT+tf/j Mouse. Front. Physiol. 2016, 7, 654.
- Haces, M.L.; Hernández-Fonseca, K.; Medina-Campos, O.N.; Montiel, T.; Pedraza-Chaverri, J.; Massieu, L. Antioxidant Capacity Contributes to Protection of Ketone Bodies against Oxidative Damage Induced during Hypoglycemic Conditions. Exp. Neurol. 2008, 211, 85–96.
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, Ketone Bodies, and Mitochondrial Energy Transduction. FASEB J. 1995, 9, 651–658.
- Veech, R.L. The Determination of the Redox States and Phosphorylation Potential in Living Tissues and Their Relationship to Metabolic Control of Disease Phenotypes. Biochem. Mol. Biol. Educ. 2006, 34, 168–179.
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a Dietary Ketone Ester on Hippocampal Glycolytic and Tricarboxylic Acid Cycle Intermediates and Amino Acids in a 3xTgAD Mouse Model of Alzheimer’s Disease. J. Neurochem. 2017, 141, 195–207.
- Xin, L.; Ipek, Ö.; Beaumont, M.; Shevlyakova, M.; Christinat, N.; Masoodi, M.; Greenberg, N.; Gruetter, R.; Cuenoud, B. Nutritional Ketosis Increases NAD+/NADH Ratio in Healthy Human Brain: An in Vivo Study by 31P-MRS. Front. Nutr. 2018, 5, 62.
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 Mediates Neuroprotection of Ketones against Ischemic Stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1783–1789.
- Yin, J.; Nielsen, M.; Li, S.; Shi, J. Ketones Improves Apolipoprotein E4-Related Memory Deficiency via Sirtuin 3. Aging 2019, 11, 4579–4586.
- Kashiwaya, Y.; King, M.T.; Veech, R.L. Substrate Signaling by Insulin. Am. J. Cardiol. 1997, 80, 50A–64A.
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones Inhibit Mitochondrial Production of Reactive Oxygen Species Production Following Glutamate Excitotoxicity by Increasing NADH Oxidation. Neuroscience 2007, 145, 256–264.
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate Regulates Energy Metabolism and Induces BDNF Expression in Cerebral Cortical Neurons. J. Neurochem. 2016, 139, 769–781.
- Pasyukova, E.G.; Vaiserman, A.M. HDAC Inhibitors: A New Promising Drug Class in Anti-Aging Research. Mech. Ageing Dev. 2017, 166, 6–15.
- Wang, X.; Wu, X.; Liu, Q.; Kong, G.; Zhou, J.; Jiang, J.; Wu, X.; Huang, Z.; Su, W.; Zhu, Q. Ketogenic Metabolism Inhibits Histone Deacetylase (HDAC) and Reduces Oxidative Stress After Spinal Cord Injury in Rats. Neuroscience 2017, 366, 36–43.
- Li, B.; Yu, Y.; Liu, K.; Zhang, Y.; Geng, Q.; Zhang, F.; Li, Y.; Qi, J. β-Hydroxybutyrate Inhibits Histone Deacetylase 3 to Promote Claudin-5 Generation and Attenuate Cardiac Microvascular Hyperpermeability in Diabetes. Diabetologia 2021, 64, 226–239.
- Guerrieri, F.; Nicoletti, C.; Adorisio, E.; Caraccio, G.; Leonetti, P.; Zanotti, F.; Cantatore, P. Correlation between Decreased Expression of Mitochondrial F0F1-ATP Synthase and Low Regenerating Capability of the Liver after Partial Hepatectomy in Hypothyroid Rats. J. Bioenerg. Biomembr. 2000, 32, 183–191.
- Mashek, D.G. Hepatic Fatty Acid Trafficking: Multiple Forks in the Road. Adv. Nutr. 2013, 4, 697–710.
- Barrows, B.R.; Parks, E.J. Contributions of Different Fatty Acid Sources to Very Low-Density Lipoprotein-Triacylglycerol in the Fasted and Fed States. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452.
- Passarella, S.; de Bari, L.; Valenti, D.; Pizzuto, R.; Paventi, G.; Atlante, A. Mitochondria and L-Lactate Metabolism. FEBS Lett. 2008, 582, 3569–3576.
- Paventi, G.; Pizzuto, R.; Passarella, S. The Occurrence of L-Lactate Dehydrogenase in the Inner Mitochondrial Compartment of Pig Liver. Biochem. Biophys. Res. Commun. 2017, 489, 255–261.
- Fromenty, B.; Roden, M. Mitochondrial Alterations in Fatty Liver Diseases. J. Hepatol. 2023, 78, 415–429.
- Zhang, C.; Zhao, Y.; Yu, M.; Qin, J.; Ye, B.; Wang, Q. Mitochondrial Dysfunction and Chronic Liver Disease. Curr. Issues Mol. Biol. 2022, 44, 3156–3165.
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375.
- Grattagliano, I.; de Bari, O.; Bernardo, T.C.; Oliveira, P.J.; Wang, D.Q.-H.; Portincasa, P. Role of Mitochondria in Nonalcoholic Fatty Liver Disease-from Origin to Propagation. Clin. Biochem. 2012, 45, 610–618.
- Muriel, P.; Gordillo, K.R. Role of Oxidative Stress in Liver Health and Disease. Oxid. Med. Cell. Longev. 2016, 2016, 9037051.
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, Oxidative Stress and Cell Death. Apoptosis 2007, 12, 913–922.
- Simões, I.C.M.; Fontes, A.; Pinton, P.; Zischka, H.; Wieckowski, M.R. Mitochondria in Non-Alcoholic Fatty Liver Disease. Int. J. Biochem. Cell Biol. 2018, 95, 93–99.
- Zuo, J.; Zhang, Z.; Luo, M.; Zhou, L.; Nice, E.C.; Zhang, W.; Wang, C.; Huang, C. Redox Signaling at the Crossroads of Human Health and Disease. MedComm 2022, 3, e127.
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124.
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple Hits, Including Oxidative Stress, as Pathogenesis and Treatment Target in Non-Alcoholic Steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728.
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of Oxidative Stress in the Pathogenesis of Nonalcoholic Steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69.
- Figueira, T.R.; Barros, M.H.; Camargo, A.A.; Castilho, R.F.; Ferreira, J.C.B.; Kowaltowski, A.J.; Sluse, F.E.; Souza-Pinto, N.C.; Vercesi, A.E. Mitochondria as a Source of Reactive Oxygen and Nitrogen Species: From Molecular Mechanisms to Human Health. Antioxid. Redox Signal. 2013, 18, 2029–2074.
- Starkov, A.A. The Role of Mitochondria in Reactive Oxygen Species Metabolism and Signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52.
- Zhang, Y.; Wong, H.S. Are Mitochondria the Main Contributor of Reactive Oxygen Species in Cells? J. Exp. Biol. 2021, 224, jeb221606.
- del Río, L.A.; López-Huertas, E. ROS Generation in Peroxisomes and Its Role in Cell Signaling. Plant Cell Physiol. 2016, 57, pcw076.
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462.
- Zheng, W.; Sun, Q.; Li, L.; Cheng, Y.; Chen, Y.; Lv, M.; Xiang, X. Role of Endoplasmic Reticulum Stress in Hepatic Glucose and Lipid Metabolism and Therapeutic Strategies for Metabolic Liver Disease. Int. Immunopharmacol. 2022, 113, 109458.
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540.
- Matuz-Mares, D.; Vázquez-Meza, H.; Vilchis-Landeros, M.M. NOX as a Therapeutic Target in Liver Disease. Antioxidants 2022, 11, 2038.
- de Medeiros, I.C.; de Lima, J.G. Is Nonalcoholic Fatty Liver Disease an Endogenous Alcoholic Fatty Liver Disease?—A Mechanistic Hypothesis. Med. Hypotheses 2015, 85, 148–152.
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity during Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10.
- Parra-Vargas, M.; Rodriguez-Echevarria, R.; Jimenez-Chillaron, J.C. Nutritional Approaches for the Management of Nonalcoholic Fatty Liver Disease: An Evidence-Based Review. Nutrients 2020, 12, 3860.
- Mooli, R.G.R.; Ramakrishnan, S.K. Emerging Role of Hepatic Ketogenesis in Fatty Liver Disease. Front. Physiol. 2022, 13, 946474.
- Sripongpun, P.; Churuangsuk, C.; Bunchorntavakul, C. Current Evidence Concerning Effects of Ketogenic Diet and Intermittent Fasting in Patients with Nonalcoholic Fatty Liver. J. Clin. Transl. Hepatol. 2022, 10, 730–739.
- Watanabe, M.; Tozzi, R.; Risi, R.; Tuccinardi, D.; Mariani, S.; Basciani, S.; Spera, G.; Lubrano, C.; Gnessi, L. Beneficial Effects of the Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Comprehensive Review of the Literature. Obes. Rev. 2020, 21, e13024.
- Tendler, D.; Lin, S.; Yancy, W.S.; Mavropoulos, J.; Sylvestre, P.; Rockey, D.C.; Westman, E.C. The Effect of a Low-Carbohydrate, Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Pilot Study. Dig. Dis. Sci. 2007, 52, 589–593.
- Paoli, A.; Tinsley, G.; Bianco, A.; Moro, T. The Influence of Meal Frequency and Timing on Health in Humans: The Role of Fasting. Nutrients 2019, 11, 719.