 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simone Allegrini | -- | 1218 | 2023-07-17 12:09:06 | | | |
| 2 | Catherine Yang | Meta information modification | 1218 | 2023-07-18 03:29:52 | | |
Video Upload Options
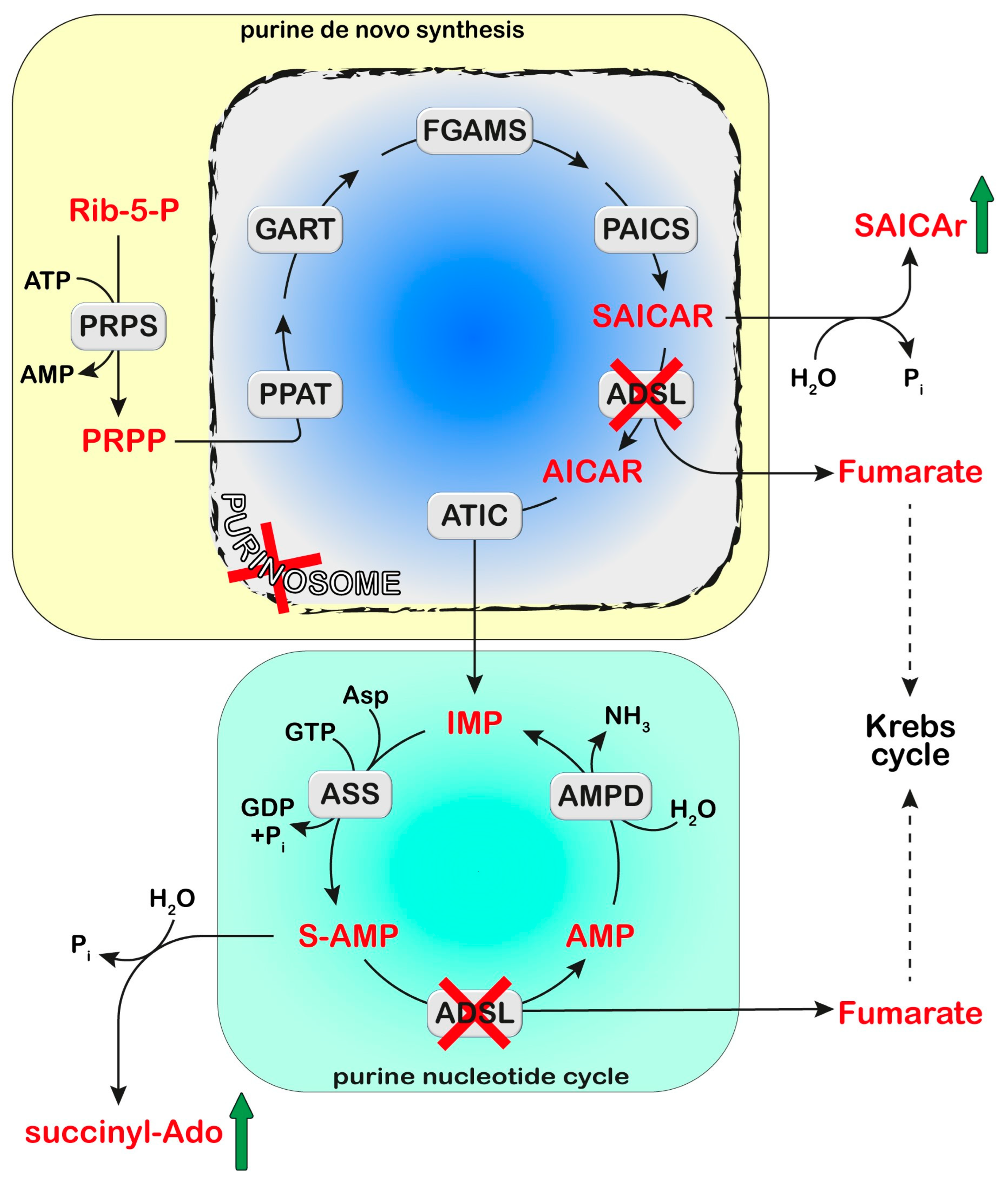
Adenylosuccinate Lyase (ADSL) is a homotetrameric enzyme exhibiting a dual catalytic role: the conversion of succinylaminoimidazolecarboxamide (SAICA)-ribotide (SAICAR) into AICA-ribotide (AICAR) (de novo purine synthesis pathway) and the formation of AMP from adenylosuccinate in the purine nucleotide cycle. ADSL deficiency is a rare autosomal recessive disorder, first described by Jaeken and Van den Berghe, caused by more than 150 different mutations (most of which missense), in the ADSL gene. In all cases, the mutations lead to an ADSL enzyme that retains some residual activity, possibly because a complete loss of activity is probably lethal in humans. The clinical presentation includes neurologic symptoms, namely intellectual disability, autism spectrum disorder, microcephaly, and seizures. Three different phenotypes have been reported on the basis of the age of onset and the severity of symptoms: the fatal neonatal form, presenting with hypokinesia, intractable seizures, and respiratory failure; the type I form presenting within the first months of life, characterized by severe psychomotor retardation, microcephaly, seizures, and autistic features; and the type II form, presenting within the first years of life, with moderate or slight psychomotor retardation]. Life expectation in ADSL deficiency is variable. The neonatal form may lead to early death, whereas onset in early childhood usually results in a stable course.
1. Diagnosis and Treatment
Cellular purine nucleotides derive mainly from de novo synthesis or nucleic acid turnover and, only marginally, from dietary intake. They are subjected to catabolism, eventually forming uric acid in humans, while bases and nucleosides may be converted back to nucleotides through the salvage pathways. ADSL deficiency is usually diagnosed, using HPLC and tandem mass spectrometry, by the detection in extracellular fluids of SAICA-riboside (SAICAr) and succinyladenosine (S-Ado), the dephosphorylated forms of the two substrates of ADSL (Figure 1). Enzyme assay in erythrocyte lysates was not completely reliable due to the tissue heterogeneity of the ADSL defect [1] and residual activity (>2% of normal) could be detected in the lymphocytes or cultured fibroblasts of patients presenting a lethal fetal and early neonatal form of ADSL deficiency [2]. More recently, whole-exome sequencing analysis has become a clinical practice [3][4]. Magnetic resonance imaging can be useful for the clinical diagnosis and for monitoring the progression of the disorder [5]. The severity of the clinical symptoms appears to correlate with the stability of the mutated enzyme and its residual activity [6][7][8]. The ratio of S-Ado/SAICAr, rather than their absolute concentrations, correlates with the severity of the phenotype [9] being less than one in the fatal form, close to one in the severe type I form, and more than one in the moderate type II form. These findings suggest that SAICAr might be the neurotoxic compound, and that S-Ado might counteract its noxious effects, as confirmed in studies conducted in experimental animals and cultured cells [10][11].
2. Proposed Mechanistic Basis
References
- Salerno, C.; Crifo, C.; Giardini, O. Adenylosuccinase deficiency: A patient with impaired erythrocyte activity and anomalous response to intravenous fructose. J. Inherit. Metab. Dis. 1995, 18, 602–608.
- Mouchegh, K.; Zikanova, M.; Hoffmann, G.F.; Kretzschmar, B.; Kuhn, T.; Mildenberger, E.; Stoltenburg-Didinger, G.; Krijt, J.; Dvorakova, L.; Honzik, T.; et al. Lethal fetal and early neonatal presentation of adenylosuccinate lyase deficiency: Observation of 6 patients in 4 families. J. Pediatr. 2007, 150, 57–61.
- Mastrogiorgio, G.; Macchiaiolo, M.; Buonuomo, P.S.; Bellacchio, E.; Bordi, M.; Vecchio, D.; Brown, K.P.; Watson, N.K.; Contardi, B.; Cecconi, F.; et al. Clinical and molecular characterization of patients with adenylosuccinate lyase deficiency. Orphanet J. Rare Dis. 2021, 16, 112.
- Mao, X.; Li, K.; Tang, B.; Luo, Y.; Ding, D.; Zhao, Y.; Wang, C.; Zhou, X.; Liu, Z.; Zhang, Y.; et al. Novel mutations in ADSL for Adenylosuccinate Lyase Deficiency identified by the combination of Trio-WES and constantly updated guidelines. Sci. Rep. 2017, 7, 1625.
- Jurecka, A.; Jurkiewicz, E.; Tylki-Szymanska, A. Magnetic resonance imaging of the brain in adenylosuccinate lyase deficiency: A report of seven cases and a review of the literature. Eur. J. Pediatr. 2012, 171, 131–138.
- Zikanova, M.; Skopova, V.; Hnizda, A.; Krijt, J.; Kmoch, S. Biochemical and structural analysis of 14 mutant adsl enzyme complexes and correlation to phenotypic heterogeneity of adenylosuccinate lyase deficiency. Hum. Mutat. 2010, 31, 445–455.
- Ray, S.P.; Duval, N.; Wilkinson, T.G., 2nd; Shaheen, S.E.; Ghosh, K.; Patterson, D. Inherent properties of adenylosuccinate lyase could explain S-Ado/SAICAr ratio due to homozygous R426H and R303C mutations. Biochim. Biophys. Acta 2013, 1834, 1545–1553.
- Kmoch, S.; Hartmannova, H.; Stiburkova, B.; Krijt, J.; Zikanova, M.; Sebesta, I. Human adenylosuccinate lyase (ADSL), cloning and characterization of full-length cDNA and its isoform, gene structure and molecular basis for ADSL deficiency in six patients. Hum. Mol. Genet. 2000, 9, 1501–1513.
- Van den Bergh, F.; Vincent, M.F.; Jaeken, J.; Van den Berghe, G. Residual adenylosuccinase activities in fibroblasts of adenylosuccinase-deficient children: Parallel deficiency with adenylosuccinate and succinyl-AICAR in profoundly retarded patients and non-parallel deficiency in a mildly retarded girl. J. Inherit. Metab. Dis. 1993, 16, 415–424.
- Stone, T.W.; Roberts, L.A.; Morris, B.J.; Jones, P.A.; Ogilvy, H.A.; Behan, W.M.; Duley, J.A.; Simmonds, H.A.; Vincent, M.F.; van den Berghe, G. Succinylpurines induce neuronal damage in the rat brain. Adv. Exp. Med. Biol. 1998, 431, 185–189.
- Souckova, O.; Skopova, V.; Baresova, V.; Sedlak, D.; Bleyer, A.J.; Kmoch, S.; Zikanova, M. Metabolites of De Novo Purine Synthesis: Metabolic Regulators and Cytotoxic Compounds. Metabolites 2022, 12, 1210.
- Jurecka, A.; Zikanova, M.; Kmoch, S.; Tylki-Szymanska, A. Adenylosuccinate lyase deficiency. J. Inherit. Metab. Dis. 2015, 38, 231–242.
- Lefevre, F.; Aronson, N. Ketogenic diet for the treatment of refractory epilepsy in children: A systematic review of efficacy. Pediatrics 2000, 105, E46.
- Shehata, N.I.; Abdelsamad, M.A.; Amin, H.A.A.; Sadik, N.A.H.; Shaheen, A.A. Ameliorating effect of ketogenic diet on acute status epilepticus: Insights into biochemical and histological changes in rat hippocampus. J. Food Biochem. 2022, 46, e14217.
- Jurecka, A.; Opoka-Winiarska, V.; Rokicki, D.; Tylki-Szymanska, A. Neurologic presentation, diagnostics, and therapeutic insights in a severe case of adenylosuccinate lyase deficiency. J. Child Neurol. 2012, 27, 645–649.
- Salerno, C.; D’Eufemia, P.; Finocchiaro, R.; Celli, M.; Spalice, A.; Iannetti, P.; Crifo, C.; Giardini, O. Effect of D-ribose on purine synthesis and neurological symptoms in a patient with adenylosuccinase deficiency. Biochim. Biophys. Acta 1999, 1453, 135–140.
- Jurecka, A.; Tylki-Szymanska, A.; Zikanova, M.; Krijt, J.; Kmoch, S. D-ribose therapy in four Polish patients with adenylosuccinate lyase deficiency: Absence of positive effect. J. Inherit. Metab. Dis. 2008, 31 (Suppl. S2), S329–S332.
- Van den Bergh, F.; Vincent, M.F.; Jaeken, J.; Van den Berghe, G. Functional studies in fibroblasts of adenylosuccinase-deficient children. J. Inherit. Metab. Dis. 1993, 16, 425–434.
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 2008, 320, 103–106.
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154.
- Baresova, V.; Skopova, V.; Sikora, J.; Patterson, D.; Sovova, J.; Zikanova, M.; Kmoch, S. Mutations of ATIC and ADSL affect purinosome assembly in cultured skin fibroblasts from patients with AICA-ribosiduria and ADSL deficiency. Hum. Mol. Genet. 2012, 21, 1534–1543.
- Keller, K.E.; Tan, I.S.; Lee, Y.S. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science 2012, 338, 1069–1072.
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980.
- Dutto, I.; Gerhards, J.; Herrera, A.; Souckova, O.; Skopova, V.; Smak, J.A.; Junza, A.; Yanes, O.; Boeckx, C.; Burkhalter, M.D.; et al. Pathway-specific effects of ADSL deficiency on neurodevelopment. elife 2022, 11, e70518.
- Bruce-Gregorios, J.H.; Agarwal, R.P.; Oracion, A.; Ramirez, A.; Lin, L. Effects of methotrexate on RNA and purine synthesis of astrocytes in primary culture. J. Neuropathol. Exp. Neurol. 1991, 50, 770–778.

