Adenylosuccinate Lyase (ADSL) is a homotetrameric enzyme exhibiting a dual catalytic role: the conversion of succinylaminoimidazolecarboxamide (SAICA)-ribotide (SAICAR) into AICA-ribotide (AICAR) (de novo purine synthesis pathway) and the formation of AMP from adenylosuccinate in the purine nucleotide cycle. ADSL deficiency is a rare autosomal recessive disorder, first described by Jaeken and Van den Berghe, caused by more than 150 different mutations (most of which missense), in the ADSL gene. In all cases, the mutations lead to an ADSL enzyme that retains some residual activity, possibly because a complete loss of activity is probably lethal in humans. The clinical presentation includes neurologic symptoms, namely intellectual disability, autism spectrum disorder, microcephaly, and seizures. Three different phenotypes have been reported on the basis of the age of onset and the severity of symptoms: the fatal neonatal form, presenting with hypokinesia, intractable seizures, and respiratory failure; the type I form presenting within the first months of life, characterized by severe psychomotor retardation, microcephaly, seizures, and autistic features; and the type II form, presenting within the first years of life, with moderate or slight psychomotor retardation]. Life expectation in ADSL deficiency is variable. The neonatal form may lead to early death, whereas onset in early childhood usually results in a stable course.
1. Diagnosis and Treatment
Cellular purine nucleotides derive mainly from de novo synthesis or nucleic acid turnover and, only marginally, from dietary intake. They are subjected to catabolism, eventually forming uric acid in humans, while bases and nucleosides may be converted back to nucleotides through the salvage pathways. ADSL deficiency is usually diagnosed, using HPLC and tandem mass spectrometry, by the detection in extracellular fluids of SAICA-riboside (SAICAr) and succinyladenosine (S-Ado), the dephosphorylated forms of the two substrates of ADSL (Figure 2). Enzyme assay in erythrocyte lysates was not completely reliable due to the tissue heterogeneity of the ADSL defect [87] and residual activity (>2% of normal) could be detected in the lymphocytes or cultured fibroblasts of patients presenting a lethal fetal and early neonatal form of ADSL deficiency [88]. More recently, whole-exome sequencing analysis has become a clinical practice [85,89]. Magnetic resonance imaging can be useful for the clinical diagnosis and for monitoring the progression of the disorder [90]. The severity of the clinical symptoms appears to correlate with the stability of the mutated enzyme and its residual activity [91,92,93]. The ratio of S-Ado/SAICAr, rather than their absolute concentrations, correlates with the severity of the phenotype [94] being less than one in the fatal form, close to one in the severe type I form, and more than one in the moderate type II form. These findings suggest that SAICAr might be the neurotoxic compound, and that S-Ado might counteract its noxious effects, as confirmed in studies conducted in experimental animals and cultured cells [95,96].
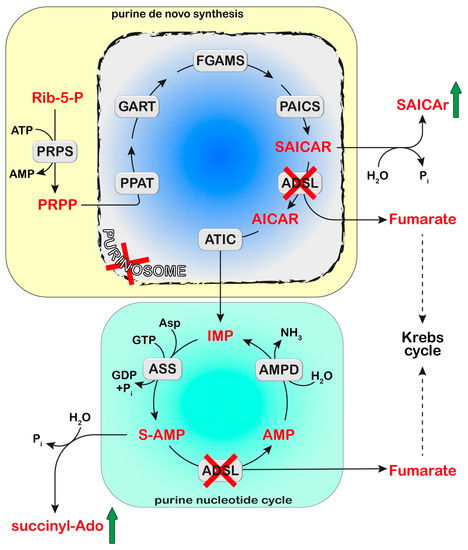
Figure 2. Adenylosuccinate lyase (ADSL) is involved both in the de novo purine synthesis (yellow box) and the purine nucleotide cycle (green box). The deficiency of ADSL causes an accumulation of its substrates, succinyl aminoimidazole carboxamide ribotide (SAICAR) and succinyl-AMP (S-AMP), which are both dephosphorylated and converted to SAICA-riboside (SAICAr) and succinyl-Ado (S-Ado), respectively. The formation of the purinosome complex (blue box) is impaired in cases of ADSL deficiency. Phosphoribosylpyrophosphate (PRPP) is synthesized from ribose-5-phosphate (Rib-5-P) by PRPP synthetase (PRPS). Six enzymes can form the purinosome and catalyze the ten steps required to convert PRPP into IMP: PRPP amidotransferase (PPAT), trifunctional phosphoribosyl glicinamide synthetase/phosphoribosyl glycinamide transformylase/phophoribosyl aminoimidazole synthetase (GART), phosphoribosyl glycinamidine synthase (FGAMS), bifunctional phosphoribosyl aminoimidazole carboxylase/phosphoribosyl aminoimidazole succinocarboxamide synthetase (PAICS), ADSL, and bifunctional 5-aminoimidazole-4-carboxamide ribonucleotide transformylase/IMP cyclohydrolase (ATIC). IMP enters the purine nucleotide cycle composed of adenylosuccinate synthase (ASS), ADSL, and AMP deaminase (AMPD). Asp: Aspartate.
No effective treatment is currently available for ADSL deficiency. The therapeutic approach with anticonvulsive drugs is primarily aimed at controlling seizure frequency with minimal side effects [
86]. A ketogenic diet has been proposed for the treatment of refractory epilepsy [
97,
98] and used as a therapeutic tool in several cases of severe ADSL deficiency [
89,
99]. However, the beneficial effects appeared to be transitory. Treatment with D-ribose was also recommended for the therapy of ADSL deficiency [
100]. However, inconsistent effects of the treatment have been reported [
89,
100,
101]. Cultured fibroblasts from ADSL-deficient patients exhibit normal purine nucleotide levels, growth rates, and ATP concentrations, thus suggesting that the symptoms of ADSL deficiency are caused by the accumulation of succinylpurines, rather than by the intracellular deficiency of purine nucleotides [
102].
2. Proposed Mechanistic Basis
ADSL is a component of the purinosome complex, composed of six enzymes catalyzing the ten chemical steps necessary to convert PRPP into IMP [
103] (
Figure 2). The assembly occurs in the cytosol upon the depletion or increased demand for purines [
1]. Using confocal microscopy, Baresova et al. [
104] demonstrated that purinosome assembly in the skin fibroblasts of ADSL-deficient patients, cultured in a purine-depleted medium, was significantly impaired and the extent of the enzyme assembly correlated with the severity of the phenotype of ADSL deficiency. The impairment in purinosome assembly reduces metabolite flux through purine de novo synthesis and purine nucleotide recycling in case of a need for purine synthesis; however, the molecular signaling mechanisms that link purinosome formation impairment to clinical outcomes of ADSL deficiency still need to be elucidated.
In the past years, the role of SAICAR in perturbing glucose metabolism has been proposed. In fact, under conditions of limiting glucose, SAICAR has been shown to activate pyruvate kinase M2 (PKM2) [
105], the isoform present in cancer, but also in embryonic cells [
106]. Therefore, the accumulation of SAICAR, leading to alterations in the activity of PKM2 during development, might contribute to defects in brain maturation in ADSL-deficient subjects. However, how a perturbation in glucose metabolism might reflect in clinical outcomes of ADSL deficiency still needs to be unraveled.
Recently, a step forwards for the understanding of the molecular mechanisms underlying the clinical manifestations of ADSL deficiency has been made using a cell model in which ADSL depletion was obtained by a pool of siRNA in hTERT-immortalized human retinal cells (RPE-1 cells) [
107]. The depletion of ADSL caused a p53-dependent proliferation arrest, with no effect on cell death or senescence, and mild DNA damage, both independent of the modulation of purine de novo synthesis. DNA damage could be rescued by the addition of adenosine, thus implicating defects in the purine nucleotide cycle. This observation was sustained in vivo, in the chicken embryo system, where ADSL depletion, obtained by the silencing of the gene, caused the same effect on proliferation and growth, leading to impaired neurogenesis [
107]. Dutto et al. [
107] also demonstrated that ADSL depletion in RPE-1 cells led to impairments in ciliogenesis. Ciliary defects, leading to microcephaly, were also observed in vivo using a zebrafish model, in which the
Adsl gene was silenced with antisense morpholino oligonucleotides. These defects were rescued by treatment with methotrexate, which impairs purine de novo synthesis up- and downstream of ADSL [
108], and by the inhibition of the enzyme responsible for the formation of SAICAR (
Figure 2), but not by nucleoside supplementation. Thus, the authors concluded that the impairment in ciliogenesis was due to the accumulation of SAICAr, and therefore to a damage in the de novo synthesis, rather than to a deficiency in the purine supply [
107]. In conclusion, both defects in the purine nucleotide cycle and impaired de novo synthesis may contribute to neurodevelopmental disorders, possibly acting through specific, but unfortunately still unknown, signaling pathways.
This entry is adapted from the peer-reviewed paper 10.3390/metabo13070787