Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wang, S.; Van Schooten, F.; Jin, H.; Jonkers, D.; Godschalk, R. Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/46795 (accessed on 24 July 2026).
Wang S, Van Schooten F, Jin H, Jonkers D, Godschalk R. Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/46795. Accessed July 24, 2026.
Wang, Shan, Frederik-Jan Van Schooten, Han Jin, Daisy Jonkers, Roger Godschalk. "Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease" Encyclopedia, https://encyclopedia.pub/entry/46795 (accessed July 24, 2026).
Wang, S., Van Schooten, F., Jin, H., Jonkers, D., & Godschalk, R. (2023, July 14). Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease. In Encyclopedia. https://encyclopedia.pub/entry/46795
Wang, Shan, et al. "Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease." Encyclopedia. Web. 14 July, 2023.
Copy Citation
Evidence is emerging for the role of intestinal tryptophan metabolism in the development of inflammatory bowel disease (IBD). In order to identify the role of altered intestinal tryptophan metabolism in IBD pathogenesis, a meta-analysis of the transcriptome was performed to identify differentially expressed genes involved in the tryptophan metabolism pathways in intestinal biopsies of IBD as compared to non-IBD controls.
inflammatory bowel disease
tryptophan metabolism
transcriptome
1. Introduction
Inflammatory bowel disease (IBD) includes two phenotypes: ulcerative colitis (UC) and Crohn’s disease (CD). UC is characterized by continuous inflammation that is limited to the colon, while CD involves any part of the gastrointestinal (GI) tract in a non-continuous fashion and, unlike UC, is commonly associated with complications, such as strictures, abscesses, and fistulas. Histologically, UC shows superficial inflammatory changes limited to the mucosa and submucosa, while CD can occur in all layers of the bowel wall [1]. With an alternating relapsing-remitting disease course, the outcome of both CD and UC could vary from minor symptoms with prolonged periods of remission to active disease with recurrent exacerbations and severe life-threatening conditions that result in hospitalization, surgical intervention, or even death [2]. Current treatment strategies aim at controlling mucosal inflammation, but these are not always effective. Patients in remission, even after surgical resection, often relapse [3]. So far, the etiology of IBD remains largely unknown, though it is hypothesized that the onset is due to an aberrant intestinal immune response to environmental triggers, catalyzed by the genetic susceptibility of the individual [4]. It is of great importance to gain a better understanding of IBD pathogenesis and expand the therapeutic armamentarium.
Intestinal tryptophan (TRP) metabolism involves a complex interaction between host genetic, microbial, and dietary factors. TRP is an essential amino acid that should be ingested via TRP-rich foods, such as lean meat, fish, dairy products, nuts and seeds, and so on [5]. TRP metabolism follows three main pathways in the GI tract [6][7] (see Figure 1). Firstly, the ingested TRP can be metabolized by the kynurenine pathway (KP) via the rate-limiting enzyme indoleamine 2,3-dioxygenase 1 (IDO1), with notable expression in mucosal and immune cells. Under normal physical conditions, an intestinal KP is present with a minimal (5–10%) contribution to TRP degradation, but this contribution may increase significantly after immune activation. Secondly, about 1–2% of dietary TRP enters the serotonin (5-hydroxytryptamine, 5-HT) pathway via tryptophan hydroxylase (TPH) in the gut, mainly within the enterochromaffin cells, generating approximately 95% of the total serotonin content of the human body. Thirdly, around 4–6% of TRP enters the indole pathway in gut microbiota, which produce a range of indole metabolites. These three pathways work separately but remain tightly interconnected in affecting gut homeostasis. Many TRP metabolites produced by these pathways were reported to affect the intestinal activation of aryl hydrocarbon receptor (AhR) signaling, which is important in modulating intestinal inflammation [8]. It is therefore worthwhile to study these pathways collectively to gain a complete and thorough understanding of the involvement of TRP metabolism in gut inflammatory disorders, most notably IBD.
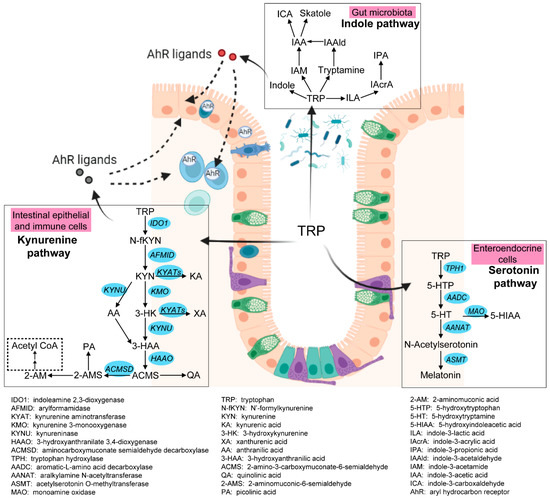
Figure 1. Three pathways of intestinal TRP metabolism. The genes involved in the kynurenine pathway and the serotonin pathway were indicated in blue. This figure was generated with BioRender (https://biorender.com/ (accessed on 29 September 2022)).
Clinical and animal experiments have identified perturbations of TRP metabolism in the development of IBD. For instance, dietary TRP deficiency enhanced dextran sodium sulfate (DSS)-induced intestinal inflammation [9], while administration of TRP or TRP metabolites might ameliorate inflammation and regulate epithelial homeostasis [10][11][12]. Decreased serum levels of TRP and increased IDO1 expression in mucosal samples were found in patients with IBD [13][14][15]. The severity of DSS-induced colitis was attenuated in TPH1−/− mice and in mice with inhibition of 5-HT synthesis, suggesting that 5-HT worsens intestinal inflammation [16]. These findings indicate that genes and metabolites involved in intestinal TRP metabolism could be potential biomarkers for intestinal inflammation and may be of interest for predicting relapse. In addition, modulation of intestinal TRP metabolism could offer potential targets for preventive and therapeutic interventions for IBD patients. So far, both TRP and its metabolite kynurenine (KYN), as well as the rate-limiting enzymes IDO1 and TPH1, have been widely studied in intestinal disorders, while the regulatory role of the other genes and metabolites within TRP metabolism pathways in IBD remains largely unexplored and thus needs to be investigated.
In order to gain a complete understanding of the three metabolic pathways of TRP in IBD, a meta-analysis of publicly available transcriptomics datasets derived from intestinal biopsies of IBD patients and non-IBD controls was performed to detect differentially expressed genes involved in the KP and serotonin pathways.
2. Meta-Analysis of Gene Expressions and Summary of Metabolites Involved in Intestinal TRP Metabolism
2.1. Decreased TRP Absorption in IBD Patients
Dietary TRP is absorbed by enterocytes apically via the B0AT1 (encoded by SLC6A19) epithelial amino acid transporter and basolaterally transported via the aromatic amino acid transporter TAT1 (SLC16A10) protein [7]. As shown in Figure 2, the expression of SLC6A19 was significantly lower in colonic biopsies of active UC and ileal biopsies of active CD. Moreover, the expression of SLC16A10 was significantly decreased only in ileal biopsies of active CD. In accordance with decreased gene expression of TRP transporters, nine out of eleven studies identified decreased levels of TRP in the serum/plasma of patients with IBD, with a stronger reduction in patients with active disease. Additionally, five out of seven studies found that the TRP concentration increased in the stool samples of patients with IBD (Table S4). Even though the TRP consumption from the diet was unknown, these results suggest that IBD patients with active disease may have decreased TRP absorption from the intestinal tract.
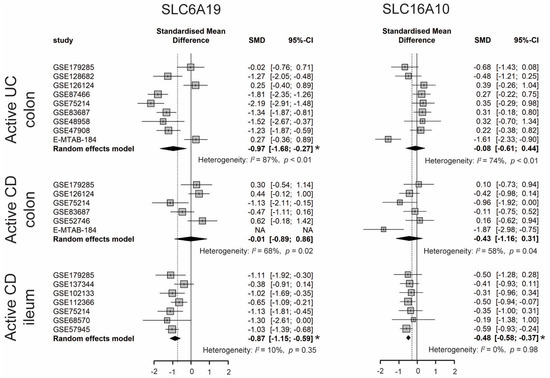
Figure 2. Forest plot for differential gene expression of SLC6A19 and SLC16A10 across studies of each IBD subtype as compared to non-IBD controls. * The p-value of the pooled effect size (ES) is less than 0.05. SMD, standardized mean difference; CI, confidence interval.
2.2. Enhanced Kynurenine Pathway (KP) in IBD Patients
Intestinal TRP metabolism through KP is mediated by the rate-limiting enzyme IDO1, which results in the production of KYN. KYN is metabolized mainly by hydroxylation to 3-hydroxykynurenine (3-HK) by kynurenine 3-monooxygenase (KMO), followed by hydrolysis of 3-HK to 3-hydroxyanthranilic acid (3-HAA) by kynureninase (KYNU). As shown in Figure 3, the expression levels of IDO1, KMO, and KYNU were significantly higher in both active CD and UC patients, indicating an enhanced KP in the gut of IBD patients. Consistently, nine out of twelve metabolomics studies identified increased KYN and/or KYN/TRP levels in either blood, stool, or colonic biopsies of IBD, with an even stronger increase in patients with active inflammation. The decreased TRP absorption and enhanced KP may synergistically contribute to the reduced blood TRP levels in IBD. When compared to inflamed colonic biopsies, the KP is less activated in inflamed ileal biopsies, as indicated by a smaller ES for IDO1, KMO, and KYNU.
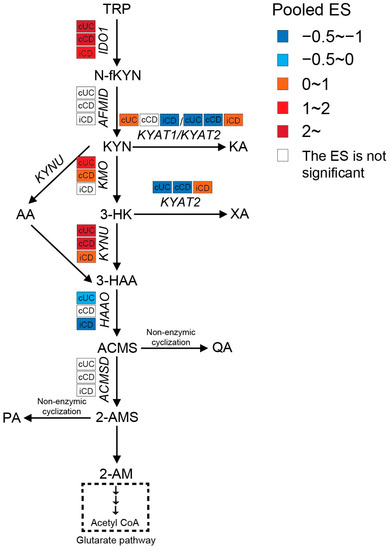
Figure 3. Pooled effect size (ES) for genes involved in KP across studies of each IBD subtype as compared to non-IBD controls. For an additional forest plot of these genes. cUC, colonic biopsies of active UC; cCD, colonic biopsies of active CD; iCD, ileal biopsies of active CD.
Both KYN and 3-HK can be transaminated by kynurenine aminotransferases (KYATs) to form kynurenic acid (KA) and xanthurenic acid (XA). These reactions by KYATs are usually of minor significance because of the high Km of their two substrates when compared to KMO and KYNU [17]. There are four KYAT isoenzymes reported, of which KYAT1 (also known as cysteine conjugate beta-lyase, CCBL1) and KYAT2 (aminoadipate aminotransferase, AADAT) play capital roles in humans. Although KYAT1 and KYAT2 possess overlapping biochemical properties, KYAT1 showed less catalytic efficiency but higher specific activity for the transformation of KYN to KA when compared to KYAT2. Moreover, KYAT1 is not actively involved in the transformation of 3-HK to XA [18]. A meta-analysis of the transcriptome showed significantly less expression of KYAT1 in inflamed ileum and less KYAT2 in inflamed colon of CD patients when compared to controls. Increased KYAT1 and decreased KYAT2 expressions were observed in colonic biopsies of UC patients. In line with gene expression, the metabolomics studies showed that serum levels of KA were lower in CD patients (especially in active status) in all included studies when compared to UC and controls, which could be a potential indicator of CD. XA also showed a decreasing trend in blood samples from IBD patients. It should be pointed out that KYAT2 was induced in inflamed ileal biopsies but inhibited in colonic biopsies from CD patients. However, none of the metabolomics studies compared the XA levels between ileal-only CD and colon-only CD, which makes it difficult to validate its role in distinguishing these two subtypes. It is worthwhile to perform further studies to confirm the difference in KYAT2 expression combined with XA concentration between ileal-only CD and colon-only CD. The differences in KA and XA between UC and controls are less clear.
The most active KP enzyme is 3-hydroxyanthranilate 3,4-dioxygenase (HAAO), which catalyzes the fast conversion of 3-HAA to 2-amino-3-carboxymuconate-6-semialdehyde (ACMS) and hence low 3-HAA levels in tissue and blood [19]. As shown in Figure 3, the gene expression of HAAO was significantly decreased in active cUC and iCD, with a stronger reduction in iCD compared to cUC. Enhanced expression of IDO1 and KYNU, but inhibition of HAAO might result in accumulation of 3-HAA. Indeed, Huhn et al. [20] observed significantly elevated levels of 3-HAA specifically in inflamed ileal biopsies from CD patients, but not in colonic samples from CD or UC patients, which might be a potential biomarker allowing the localization of the inflammation.
Subsequently, part of ACMS favors its non-enzymatic cyclization to quinolinic acid (QA). Studies with antibodies to QA demonstrated that immune cells such as mononuclear cells and tissue macrophages are the main cell types that are capable of synthesizing and storing QA. Based on previous findings, QA is strongly elevated when these cells are stimulated by immune activators such as IFN-γ [21]. Consistently, the systematic review of the metabolome also identified increased QA levels in blood, stool, and biopsy samples of IBD, especially in patients with active disease. The other two metabolic branches involve decarboxylation of ACMS by 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase (ACMSD) to form 2-aminomuconic-6-semialdehyde (2-AMS), which can further be converted to picolinic acid (PA) via non-enzymatic cyclization, to 2-aminomuconic acid (2-AM), and finally to acetyl coenzyme A (Acetyl-CoA). Acetyl-CoA feeds into the glutarate pathway to yield energy. Under physiological conditions, most TRP that enters the KP pathway is converted to ATP, CO2, and water in the glutarate pathway. PA is only produced when the flux of metabolites through the glutarate pathway is high and the enzymes of the glutarate pathway are saturated [22]. The meta-analysis of the transcriptome showed no significant difference in ACMSD expression between IBD and controls (Figure 3). However, the metabolomics studies identified either decreased or unchanged PA levels in bio-samples of IBD compared to controls. These results suggest enhanced activity of the glutarate pathway to refuel the energy deficiency of epithelial cells in inflamed intestinal biopsies from IBD patients.
KYN and most of its metabolites (including KA, XA, AA, 3-HK, 3-HAA, PA, and QA) are implicated in the regulation of the immune response. The production of KYN and its metabolites exert immunosuppressive effects mainly by inhibition of T cell function, activation of regulatory T cells, and inhibition of Natural Killer cells and antigen-presenting cells. These effects are, at least in part, mediated by the activation of the aryl hydrocarbon receptor (AhR), which has been shown to play an anti-inflammatory role in the immune response [23][24]. KA can also activate the orphan G protein-coupled receptor GPR35, which is predominantly detected in immune cells of the GI tract, to modulate intestinal inflammation [23]. The up-regulation of IDO1, KMO, and KYNU in intestinal biopsies, as well as increased levels of KYN in bio-samples of IBD patients, are therefore likely to be a negative feedback mechanism that suppresses the ongoing inflammatory response.
Intestinal TRP metabolism through the KP also yields neuroactive metabolites, among which KA and QA have been extensively studied. KA is a neuroprotective molecule that acts as an antagonist of the N-methyl-D-aspartate (NMDA), kainic acid, and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors [25]. QA can exert neurotoxic activity by acting as an NMDA receptor agonist. The systematic review of the metabolome showed decreased blood KA concentrations in CD patients and increased blood QA levels in IBD patients. The decreased KA/QA could exert an excitotoxic effect on enteric neurons, which may be involved in intestinal hypermotility [26] and malfunction of gut-brain sensory transduction [27]. Moreover, the ratio of KA to QA in the blood was reduced in patients with major depressive disorders and was negatively correlated with blood C-reactive protein (CRP) level, one of the most commonly used measures of systemic inflammation in clinical practice [28][29]. These results may particularly provide a potential mechanism underlying the entero-active effect and the occurrence of depression in IBD patients.
2.3. Increased Interstitial Serotonin Availability in IBD Patients
Serotonin synthesis in enterochromaffin (EC) cells involves the rate-limiting step where TRP is converted to 5-hydroxytryptophan (5-HTP) by tryptophan hydroxylase 1 (TPH1), followed by decarboxylation to serotonin (5-HT) by aromatic-L-amino acid decarboxylase (AADC). As shown in Figure 4, a meta-analysis of the transcriptome showed that the expression of TPH1 was significantly lower in inflamed colonic biopsies of UC patients but not in CD patients. Even though inflamed ileal biopsies from CD also expressed less TPH1, they had a smaller pooled ES. Gene expression levels of AADC were significantly decreased in both ileal and colonic biopsies of patients with active IBD, while an even stronger reduction was observed in UC patients. These results indicate the inhibition of serotonin synthesis in IBD patients with active inflammation. Colon tissue from UC patients showed stronger inhibition of the serotonin pathway when compared to CD patients.
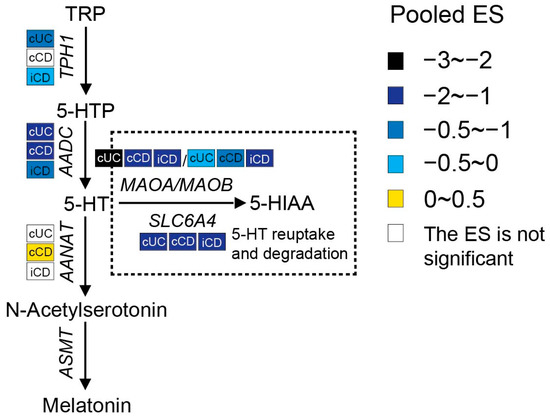
Figure 4. Pooled effect size (ES) for genes involved in the serotonin pathway across studies of each IBD subtype as compared to non-IBD controls. For additional forest plots of these genes. cUC, colonic biopsies of active UC; cCD, colonic biopsies of active CD; iCD, ileal biopsies of active CD.
Once synthesized, serotonin is rapidly packaged via the vesicular monoamine transporter into dense granules or vesicles located at the base of the cell. When EC cells are exposed to intraluminal pressure or chemical and mechanical stimulation, serotonin is released either apically to the gut lumen or basolaterally to the lamina propria. Upon its release, serotonin may take several possible routes, including having a direct influence on the gut microbiota, exerting influence on intracellular signaling by acting on 5-HT receptors (5-HTRs), or being reuptook by surrounding epithelial and immune cells via the serotonin reuptake transporter (SERT, encoded by the SLC6A4 gene), or entering the blood. In blood, serotonin is present as free serotonin or it is taken up by platelets via SERT (approximately 95%) [30][31]. Any excess serotonin in the cells is degraded by monoamine oxidase (MAO), resulting in the production of 5-hydroxyindoleacetic acid (5-HIAA), which is mainly excreted in urine. There are two forms of MAO known: MAOA and MAOB. MAOA has the highest affinity for serotonin [7]. A meta-analysis of the transcriptome showed significantly lower levels of SLC6A4 and MAO in all IBD patients with active inflammation, implying reduced reuptake and inhibited degradation of serotonin in intestinal tissues from IBD patients.
Serotonin can also be shunted into the melatonin pathways. Extra-pineal melatonin synthesis can occur in the EC cells, where serotonin can be metabolized to N-acetylserotonin by aralkylamine N-acetyltransferase (AANAT) and eventually to melatonin by acetylserotonin O-methyltransferase (ASMT). There was no obvious change in AANAT expression between IBD and controls. Moreover, ASMT was not detected in at least 80% of the included datasets, which represents the relatively low melatonin synthesis in the gut.
For the metabolites produced during this pathway, the serum level of 5-HTP was decreased in IBD identified by two studies, which is in line with reduced expression of TPH1. There was no significant difference in stool serotonin levels between IBD and control. However, four out of six studies identified increased serotonin levels in blood samples of IBD patients as compared to controls or in active CD patients as compared to inactive CD patients. Taken the transcriptome and metabolome together, these results suggest that there is an increased interstitial availability of serotonin in the lamina propria, leading to successive activation of a variety of 5-HTRs present on smooth muscles, enteric neurons, enterocytes, and immune cells to exert its biological function.
There are seven 5-HTR families identified so far, and five (5-HTR1, 5-HTR2, 5-HTR3, 5-HTR4, 5-HTR7) are expressed in the gut [32]. The conventional effects of serotonin in the gut and responding mediated receptors were summarized by Liu et al. [33] and Mawe et al. [34]. By activating these 5-HTRs, serotonin plays an important role in regulating motility, secretion, and sensory function in the gut. Furthermore, accumulating clinical and animal studies have identified the immunomodulatory role of serotonin in intestinal inflammation. Several 5-HTRs were reported to be expressed on immune cells, including B and T lymphocytes, monocytes, macrophages, and dendritic cells [35]. Through activating 5-HTRs during intestinal inflammation, serotonin can either serve as a pro-inflammatory mediator [16][36] or exert an anti-inflammatory effect on intestinal mucosa [37]. In the present study, a meta-analysis of the transcriptome showed that gene expression levels of 5-HTR3C and 5-HTR3E were significantly lower in inflamed biopsies from both CD and UC. Li et al. identified that the decreased expression of 5-HTR was the dominant mechanism underlying the desensitization of 5-HTR observed in SERT−/− mice [38]. The consistent lower levels of 5-HTR3C and 5-HTR3E among both IBD phenotypes suggests the desensitization of these receptors in response to successive potentiation of serotonin, which may reflect protective mechanisms. Additionally, the downregulation of 5-HTR1D and 5-HTR3A as well as the upregulation of 5-HTR2B were observed only in inflamed ileal biopsies from CD, and the decreased 5-HTR1E and 5-HTR4 and increased 5-HTR2A expression were specific to colonic biopsies from UC patients. The difference in 5-HTR dysregulation between IBD subtypes could at least partially explain the different clinical symptoms; it would be a potential diagnostic and therapeutic tool to improve the differentiation between IBD subtypes and, accordingly, to achieve better management of the disease. So far, there is limited research that systematically describes the 5-HTRs within the gut wall in the setting of IBD. Further human studies are required to completely understand the pathophysiological mechanisms of IBD underlying serotonin and its receptors.
References
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317.
- Law, C.C.Y.; Sasidharan, S.; Rodrigues, R.; Nguyen, D.D.; Sauk, J.; Garber, J.; Giallourakis, C.; Xavier, R.; Khalili, H.; Yajnik, V.; et al. Impact of Specialized Inpatient IBD Care on Outcomes of IBD Hospitalizations: A Cohort Study. Inflamm. Bowel Dis. 2016, 22, 2149–2157.
- Kato, J.; Yoshida, T.; Hiraoka, S. Prediction of treatment outcome and relapse in inflammatory bowel disease. Expert Rev. Clin. Immunol. 2019, 15, 667–677.
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99.
- Kaluzna-Czaplinska, J.; Gatarek, P.; Chirumbolo, S.; Chartrand, M.S.; Bjorklund, G. How important is tryptophan in human health? Crit. Rev. Food Sci. 2019, 59, 72–88.
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724.
- Keszthelyi, D.; Troost, F.J.; Masclee, A.A. Understanding the role of tryptophan and serotonin metabolism in gastrointestinal function. Neurogastroenterol. Motil. 2009, 21, 1239–1249.
- Sun, M.; Ma, N.; He, T.; Johnston, L.J.; Ma, X. Tryptophan (Trp) modulates gut homeostasis via aryl hydrocarbon receptor (AhR). Crit. Rev. Food Sci. Nutr. 2020, 60, 1760–1768.
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481.
- Islam, J.; Sato, S.; Watanabe, K.; Watanabe, T.; Ardiansyah; Hirahara, K.; Aoyama, Y.; Tomita, S.; Aso, H.; Komai, M.; et al. Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J. Nutr. Biochem. 2017, 42, 43–50.
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D'Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan Catabolites from Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity 2013, 39, 372–385.
- Wlodarska, M.; Luo, C.W.; Kolde, R.; d’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37.
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Hasler, R.; et al. Increased Tryptophan Metabolism Is Associated with Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.
- Sofia, M.A.; Ciorba, M.A.; Meckel, K.; Lim, C.K.; Guillemin, G.J.; Weber, C.R.; Bissonnette, M.; Pekow, J.R. Tryptophan Metabolism through the Kynurenine Pathway is Associated with Endoscopic Inflammation in Ulcerative Colitis. Inflamm. Bowel Dis. 2018, 24, 1471–1480.
- Gupta, N.K.; Thaker, A.I.; Kanuri, N.; Riehl, T.E.; Rowley, C.W.; Stenson, W.F.; Ciorba, M.A. Serum analysis of tryptophan catabolism pathway: Correlation with Crohn’s disease activity. Inflamm. Bowel Dis. 2012, 18, 1214–1220.
- Ghia, J.E.; Li, N.; Wang, H.Q.; Collins, M.; Deng, Y.K.; El-Sharkawy, R.T.; Cote, F.; Mallet, J.; Khan, W.I. Serotonin Has a Key Role in Pathogenesis of Experimental Colitis. Gastroenterology 2009, 137, 1649–1660.
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938.
- Rossi, F.; Miggiano, R.; Ferraris, D.M.; Rizzi, M. The Synthesis of Kynurenic Acid in Mammals: An Updated Kynurenine Aminotransferase Structural KATalogue. Front. Mol. Biosci. 2019, 6, 7.
- Badawy, A.A. Hypothesis kynurenic and quinolinic acids: The main players of the kynurenine pathway and opponents in inflammatory disease. Med. Hypotheses 2018, 118, 129–138.
- Huhn, M.; Juan, M.H.S.; Melcher, B.; Dreis, C.; Schmidt, K.G.; Schwiebs, A.; Collins, J.; Pfeilschifter, J.M.; Vieth, M.; Stein, J.; et al. Inflammation-Induced Mucosal KYNU Expression Identifies Human Ileal Crohn’s Disease. J. Clin. Med. 2020, 9, 1360.
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell Biol. 2003, 81, 247–265.
- Peters, J.C. Tryptophan Nutrition and Metabolism—An Overview. Adv. Exp. Med. Biol. 1991, 294, 345–358.
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794.
- Mandi, Y.; Vecsei, L. The kynurenine system and immunoregulation. J. Neural Transm. 2012, 119, 197–209.
- Ostapiuk, A.; Urbanska, E.M. Kynurenic acid in neurodegenerative disorders-unique neuroprotection or double-edged sword? Cns. Neurosci. Ther. 2022, 28, 19–35.
- Kaszaki, J.; Erces, D.; Varga, G.; Szabo, A.; Vecsei, L.; Boros, M. Kynurenines and intestinal neurotransmission: The role of N-methyl-d-aspartate receptors. J. Neural Transm. 2012, 119, 211–223.
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.L.; Bohorquez, D.V. A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361, eaat5236.
- Zheng, H.; Teague, T.K.; Yeh, F.C.; Burrows, K.; Figueroa-Hall, L.K.; Aupperle, R.L.; Khalsa, S.S.; Paulus, M.P.; Savitz, J. C-Reactive protein and the kynurenic acid to quinolinic acid ratio are independently associated with white matter integrity in major depressive disorder. Brain Behav. Immun. 2022, 105, 180–189.
- Savitz, J.; Drevets, W.C.; Wurfel, B.E.; Ford, B.N.; Bellgowan, P.S.F.; Victor, T.A.; Bodurka, J.; Teague, T.K.; Dantzer, R. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav. Immun. 2015, 46, 55–59.
- Tao, E.; Zhu, Z.; Hu, C.; Long, G.; Chen, B.; Guo, R.; Fang, M.; Jiang, M. Potential Roles of Enterochromaffin Cells in Early Life Stress-Induced Irritable Bowel Syndrome. Front. Cell. Neurosci. 2022, 16, 837166.
- Haq, S.; Grondin, J.A.; Khan, W.I. Tryptophan-derived serotonin-kynurenine balance in immune activation and intestinal inflammation. FASEB J. 2021, 35, e21888.
- Banskota, S.; Ghia, J.E.; Khan, W.I. Serotonin in the gut: Blessing or a curse. Biochimie 2019, 161, 56–64.
- Liu, N.; Sun, S.; Wang, P.; Sun, Y.; Hu, Q.; Wang, X. The Mechanism of Secretion and Metabolism of Gut-Derived 5-Hydroxytryptamine. Int. J. Mol. Sci. 2021, 22, 7931.
- Mawe, G.M.; Hoffman, J.M. Serotonin signalling in the gut-functions, dysfunctions and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486.
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973.
- Kim, J.J.; Wang, H.Q.; Terc, J.D.; Zambrowicz, B.; Yang, Q.M.; Khan, W.I. Blocking peripheral serotonin synthesis by telotristat etiprate (LX1032/LX1606) reduces severity of both chemical- and infection-induced intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G455–G465.
- Spohn, S.N.; Bianco, F.; Scott, R.B.; Keenan, C.M.; Linton, A.A.; O’Neill, C.H.; Bonora, E.; Dicay, M.; Lavoie, B.; Wilcox, R.L.; et al. Protective Actions of Epithelial 5-Hydroxytryptamine 4 Receptors in Normal and Inflamed Colon. Gastroenterology 2016, 151, 933–944.e3.
- Li, Q.; Wichems, C.; Heils, A.; Lesch, K.P.; Murphy, D.L. Reduction in the density and expression, but not G-protein coupling, of serotonin receptors (5-HT1A) in 5-HT transporter knock-out mice: Gender and brain region differences. J. Neurosci. 2000, 20, 7888–7895.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
940
Revisions:
2 times
(View History)
Update Date:
18 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No