Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Shan Wang and Version 2 by Dean Liu.
Evidence is emerging for the role of intestinal tryptophan metabolism in the development of inflammatory bowel disease (IBD). In order to identify the role of altered intestinal tryptophan metabolism in IBD pathogenesis, a meta-analysis of the transcriptome was performed to identify differentially expressed genes involved in the tryptophan metabolism pathways in intestinal biopsies of IBD as compared to non-IBD controls.
- inflammatory bowel disease
- tryptophan metabolism
- transcriptome
1. Introduction
Inflammatory bowel disease (IBD) includes two phenotypes: ulcerative colitis (UC) and Crohn’s disease (CD). UC is characterized by continuous inflammation that is limited to the colon, while CD involves any part of the gastrointestinal (GI) tract in a non-continuous fashion and, unlike UC, is commonly associated with complications, such as strictures, abscesses, and fistulas. Histologically, UC shows superficial inflammatory changes limited to the mucosa and submucosa, while CD can occur in all layers of the bowel wall [1]. With an alternating relapsing-remitting disease course, the outcome of both CD and UC could vary from minor symptoms with prolonged periods of remission to active disease with recurrent exacerbations and severe life-threatening conditions that result in hospitalization, surgical intervention, or even death [2]. Current treatment strategies aim at controlling mucosal inflammation, but these are not always effective. Patients in remission, even after surgical resection, often relapse [3]. So far, the etiology of IBD remains largely unknown, though it is hypothesized that the onset is due to an aberrant intestinal immune response to environmental triggers, catalyzed by the genetic susceptibility of the individual [4]. It is of great importance to gain a better understanding of IBD pathogenesis and expand the therapeutic armamentarium.
Intestinal tryptophan (TRP) metabolism involves a complex interaction between host genetic, microbial, and dietary factors. TRP is an essential amino acid that should be ingested via TRP-rich foods, such as lean meat, fish, dairy products, nuts and seeds, and so on [5]. TRP metabolism follows three main pathways in the GI tract [6][7][6,7] (see Figure 1). Firstly, the ingested TRP can be metabolized by the kynurenine pathway (KP) via the rate-limiting enzyme indoleamine 2,3-dioxygenase 1 (IDO1), with notable expression in mucosal and immune cells. Under normal physical conditions, an intestinal KP is present with a minimal (5–10%) contribution to TRP degradation, but this contribution may increase significantly after immune activation. Secondly, about 1–2% of dietary TRP enters the serotonin (5-hydroxytryptamine, 5-HT) pathway via tryptophan hydroxylase (TPH) in the gut, mainly within the enterochromaffin cells, generating approximately 95% of the total serotonin content of the human body. Thirdly, around 4–6% of TRP enters the indole pathway in gut microbiota, which produce a range of indole metabolites. These three pathways work separately but remain tightly interconnected in affecting gut homeostasis. Many TRP metabolites produced by these pathways were reported to affect the intestinal activation of aryl hydrocarbon receptor (AhR) signaling, which is important in modulating intestinal inflammation [8]. It is therefore worthwhile to study these pathways collectively to gain a complete and thorough understanding of the involvement of TRP metabolism in gut inflammatory disorders, most notably IBD.
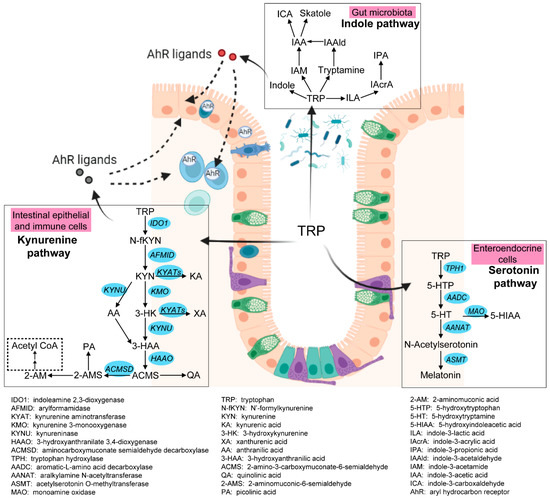
Figure 1. Three pathways of intestinal TRP metabolism. The genes involved in the kynurenine pathway and the serotonin pathway were indicated in blue. This figure was generated with BioRender (https://biorender.com/ (accessed on 29 September 2022)).
Clinical and animal experiments have identified perturbations of TRP metabolism in the development of IBD. For instance, dietary TRP deficiency enhanced dextran sodium sulfate (DSS)-induced intestinal inflammation [9], while administration of TRP or TRP metabolites might ameliorate inflammation and regulate epithelial homeostasis [10][11][12][10,11,12]. Decreased serum levels of TRP and increased IDO1 expression in mucosal samples were found in patients with IBD [13][14][15][13,14,15]. The severity of DSS-induced colitis was attenuated in TPH1−/− mice and in mice with inhibition of 5-HT synthesis, suggesting that 5-HT worsens intestinal inflammation [16]. These findings indicate that genes and metabolites involved in intestinal TRP metabolism could be potential biomarkers for intestinal inflammation and may be of interest for predicting relapse. In addition, modulation of intestinal TRP metabolism could offer potential targets for preventive and therapeutic interventions for IBD patients. So far, both TRP and its metabolite kynurenine (KYN), as well as the rate-limiting enzymes IDO1 and TPH1, have been widely studied in intestinal disorders, while the regulatory role of the other genes and metabolites within TRP metabolism pathways in IBD remains largely unexplored and thus needs to be investigated.
In order to gain a complete understanding of the three metabolic pathways of TRP in IBD, a meta-analysis of publicly available transcriptomics datasets derived from intestinal biopsies of IBD patients and non-IBD controls was performed to detect differentially expressed genes involved in the KP and serotonin pathways.
2. Meta-Analysis of Gene Expressions and Summary of Metabolites Involved in Intestinal TRP Metabolism
2.1. Decreased TRP Absorption in IBD Patients
Dietary TRP is absorbed by enterocytes apically via the B0AT1 (encoded by SLC6A19) epithelial amino acid transporter and basolaterally transported via the aromatic amino acid transporter TAT1 (SLC16A10) protein [7]. As shown in Figure 24, the expression of SLC6A19 was significantly lower in colonic biopsies of active UC and ileal biopsies of active CD. Moreover, the expression of SLC16A10 was significantly decreased only in ileal biopsies of active CD. In accordance with decreased gene expression of TRP transporters, nine out of eleven studies identified decreased levels of TRP in the serum/plasma of patients with IBD, with a stronger reduction in patients with active disease. Additionally, five out of seven studies found that the TRP concentration increased in the stool samples of patients with IBD (Table S4). Even though the TRP consumption from the diet was unknown, these results suggest that IBD patients with active disease may have decreased TRP absorption from the intestinal tract.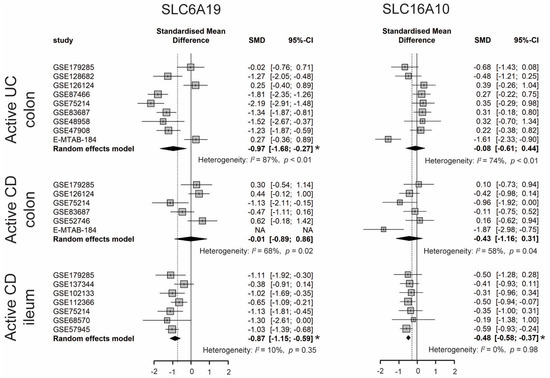
Figure 24. Forest plot for differential gene expression of SLC6A19 and SLC16A10 across studies of each IBD subtype as compared to non-IBD controls. * The p-value of the pooled effect size (ES) is less than 0.05. SMD, standardized mean difference; CI, confidence interval.
2.2. Enhanced Kynurenine Pathway (KP) in IBD Patients
Intestinal TRP metabolism through KP is mediated by the rate-limiting enzyme IDO1, which results in the production of KYN. KYN is metabolized mainly by hydroxylation to 3-hydroxykynurenine (3-HK) by kynurenine 3-monooxygenase (KMO), followed by hydrolysis of 3-HK to 3-hydroxyanthranilic acid (3-HAA) by kynureninase (KYNU). As shown in Figure 35, the expression levels of IDO1, KMO, and KYNU were significantly higher in both active CD and UC patients, indicating an enhanced KP in the gut of IBD patients. Consistently, nine out of twelve metabolomics studies identified increased KYN and/or KYN/TRP levels in either blood, stool, or colonic biopsies of IBD, with an even stronger increase in patients with active inflammation (Table S4). The decreased TRP absorption (see Section 2.1) and enhanced KP may synergistically contribute to the reduced blood TRP levels in IBD (Table S4). When compared to inflamed colonic biopsies, the KP is less activated in inflamed ileal biopsies, as indicated by a smaller ES for IDO1, KMO, and KYNU.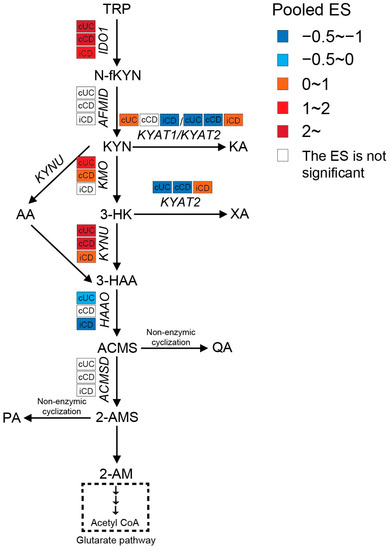
Figure 35. Pooled effect size (ES) for genes involved in KP across studies of each IBD subtype as compared to non-IBD controls. For an additional forest plot of these genes, see Supplementary Figure S1. cUC, colonic biopsies of active UC; cCD, colonic biopsies of active CD; iCD, ileal biopsies of active CD.
2.3. Increased Interstitial Serotonin Availability in IBD Patients
Serotonin synthesis in enterochromaffin (EC) cells involves the rate-limiting step where TRP is converted to 5-hydroxytryptophan (5-HTP) by tryptophan hydroxylase 1 (TPH1), followed by decarboxylation to serotonin (5-HT) by aromatic-L-amino acid decarboxylase (AADC). As shown in Figure 46, a meta-analysis of the transcriptome showed that the expression of TPH1 was significantly lower in inflamed colonic biopsies of UC patients but not in CD patients. Even though inflamed ileal biopsies from CD also expressed less TPH1, they had a smaller pooled ES. Gene expression levels of AADC were significantly decreased in both ileal and colonic biopsies of patients with active IBD, while an even stronger reduction was observed in UC patients. These results indicate the inhibition of serotonin synthesis in IBD patients with active inflammation. Colon tissue from UC patients showed stronger inhibition of the serotonin pathway when compared to CD patients.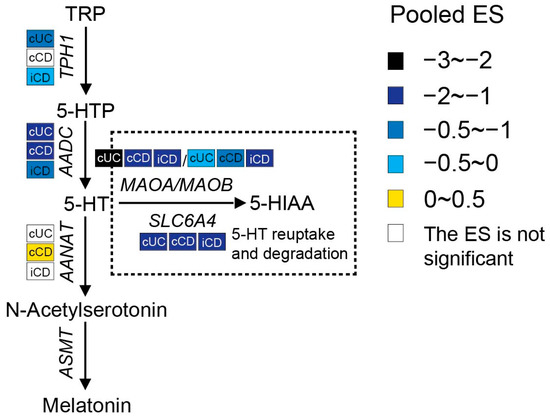
Figure 46. Pooled effect size (ES) for genes involved in the serotonin pathway across studies of each IBD subtype as compared to non-IBD controls. For additional forest plots of these genes, see Supplementary Figure S2. cUC, colonic biopsies of active UC; cCD, colonic biopsies of active CD; iCD, ileal biopsies of active CD.