 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Stanislav Rodkin | -- | 6064 | 2023-07-13 09:49:51 | | | |
| 2 | Lindsay Dong | + 4 word(s) | 6068 | 2023-07-14 05:18:59 | | |
Video Upload Options
Hydrogen sulfide (H2S) is a gaseous signaling molecule that performs various cellular functions in normal and pathological conditions. H2S has great neuroprotective potential. H2S reduces oxidative stress, lipid peroxidation, and neuroinflammation; inhibits processes associated with apoptosis, autophagy, ferroptosis and pyroptosis; prevents the destruction of the blood-brain barrier; increases the expression of neurotrophic factors; and models the activity of Ca2+ channels in neurotrauma. In addition, H2S activates neuroprotective signaling pathways in psychiatric and neurodegenerative diseases. However, high levels of H2S can cause cytotoxic effects. Thus, the development of H2S-associated neuroprotectors seems to be especially relevant.
1. Introduction
2. Classification and Molecular Mechanisms of Neurotrauma
3. Metabolism and Functions of H2S
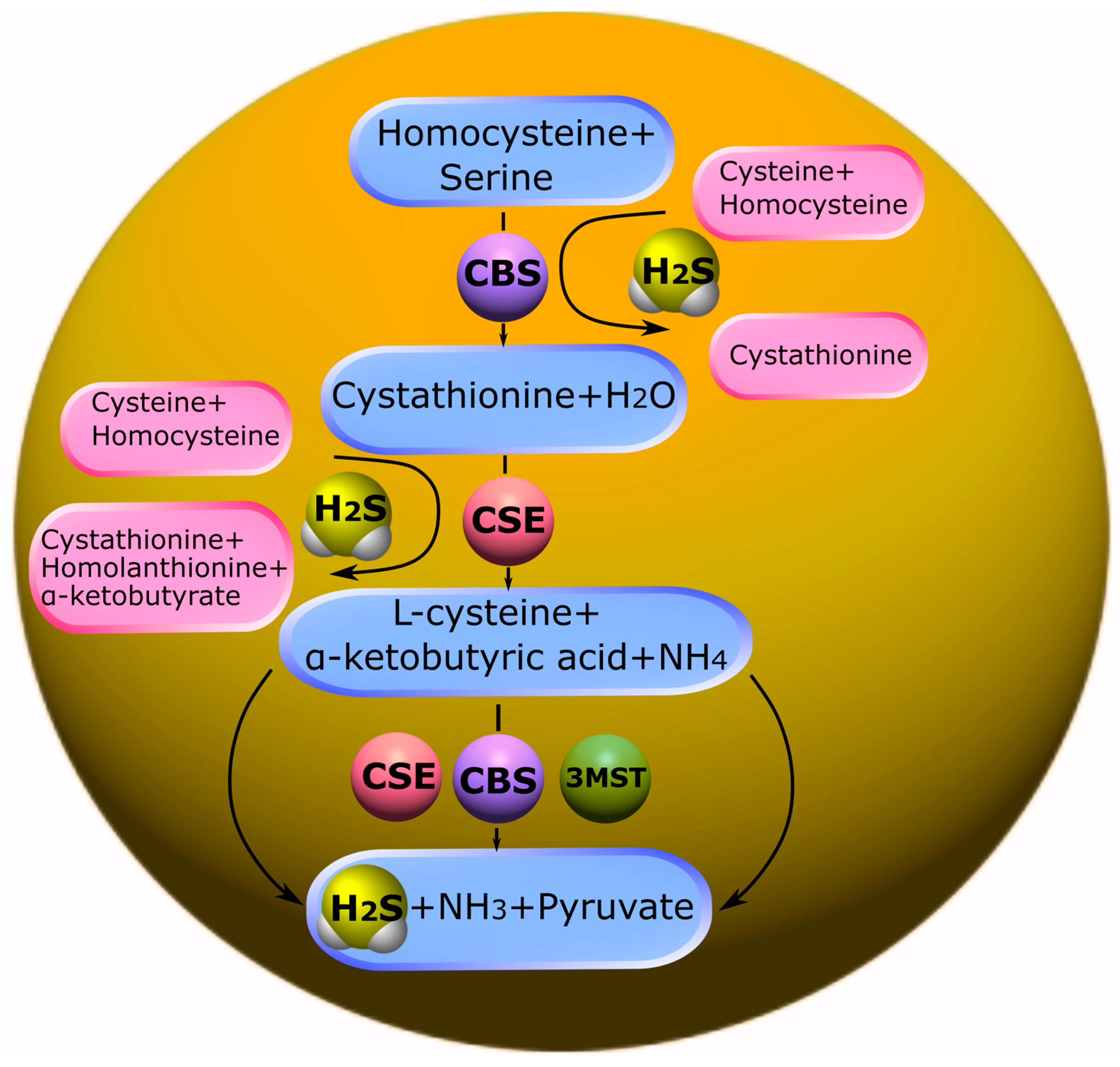
3.1. Biosynthesis of H2S and Its Deposition
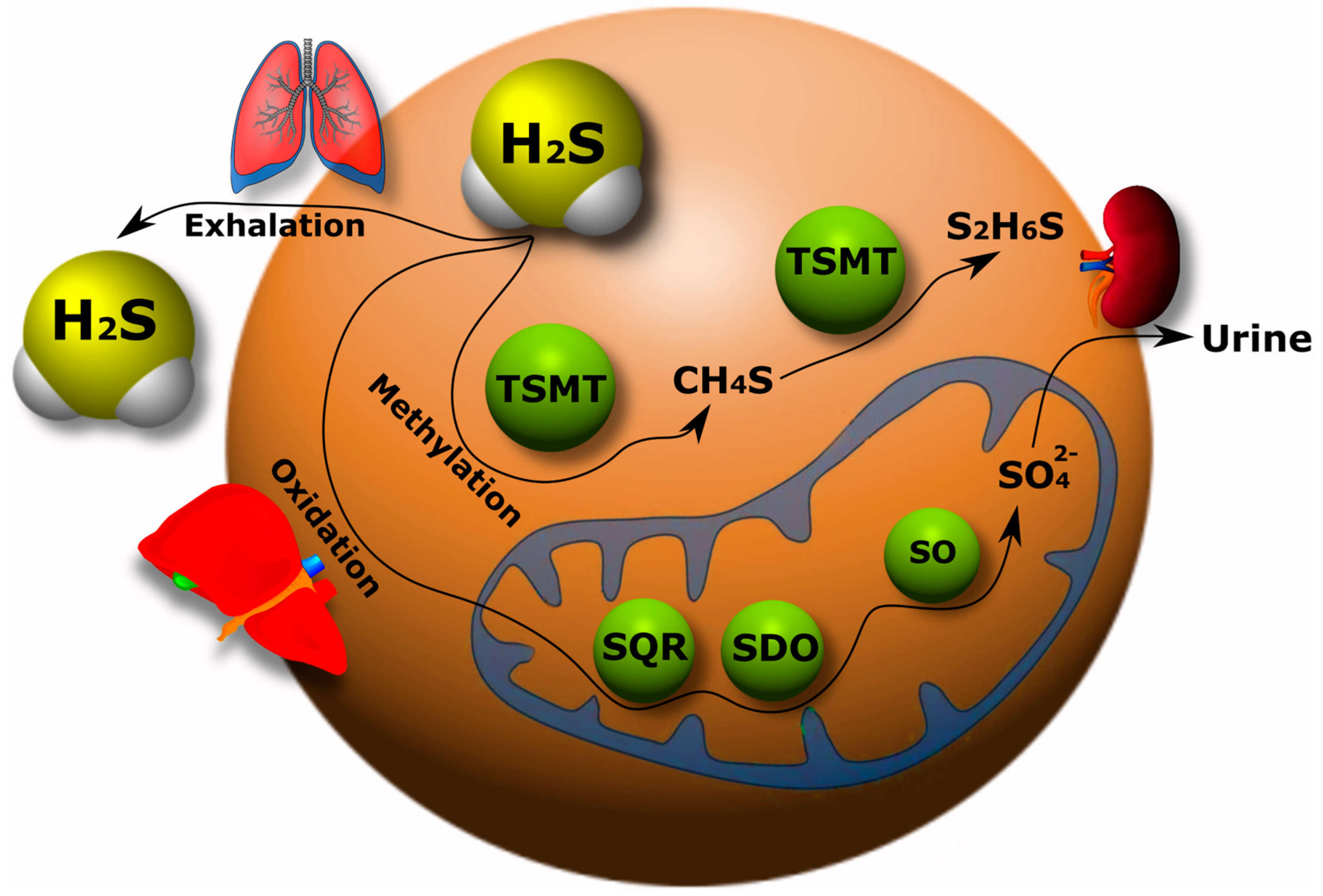
3.2. Catabolism of H2S
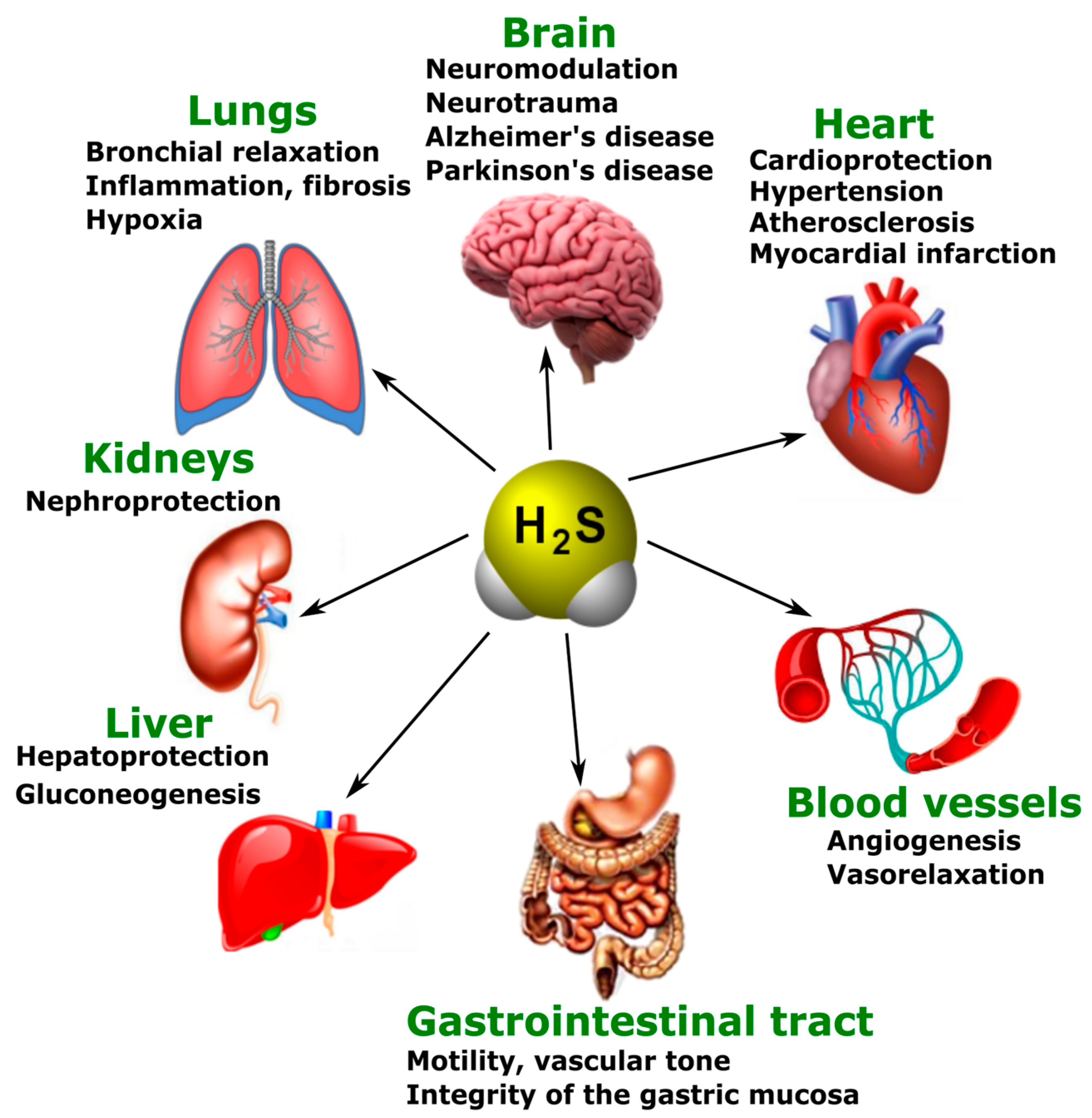
3.3. Various Biological Effects of Endogenous H2S
4. Endogenous and Exogenous H2S in Neurotrauma
4.1. Endogenous H2S Levels in Neurotrauma
4.2. Exogenous H2S: Between Neuroprotection and Neurodegeneration
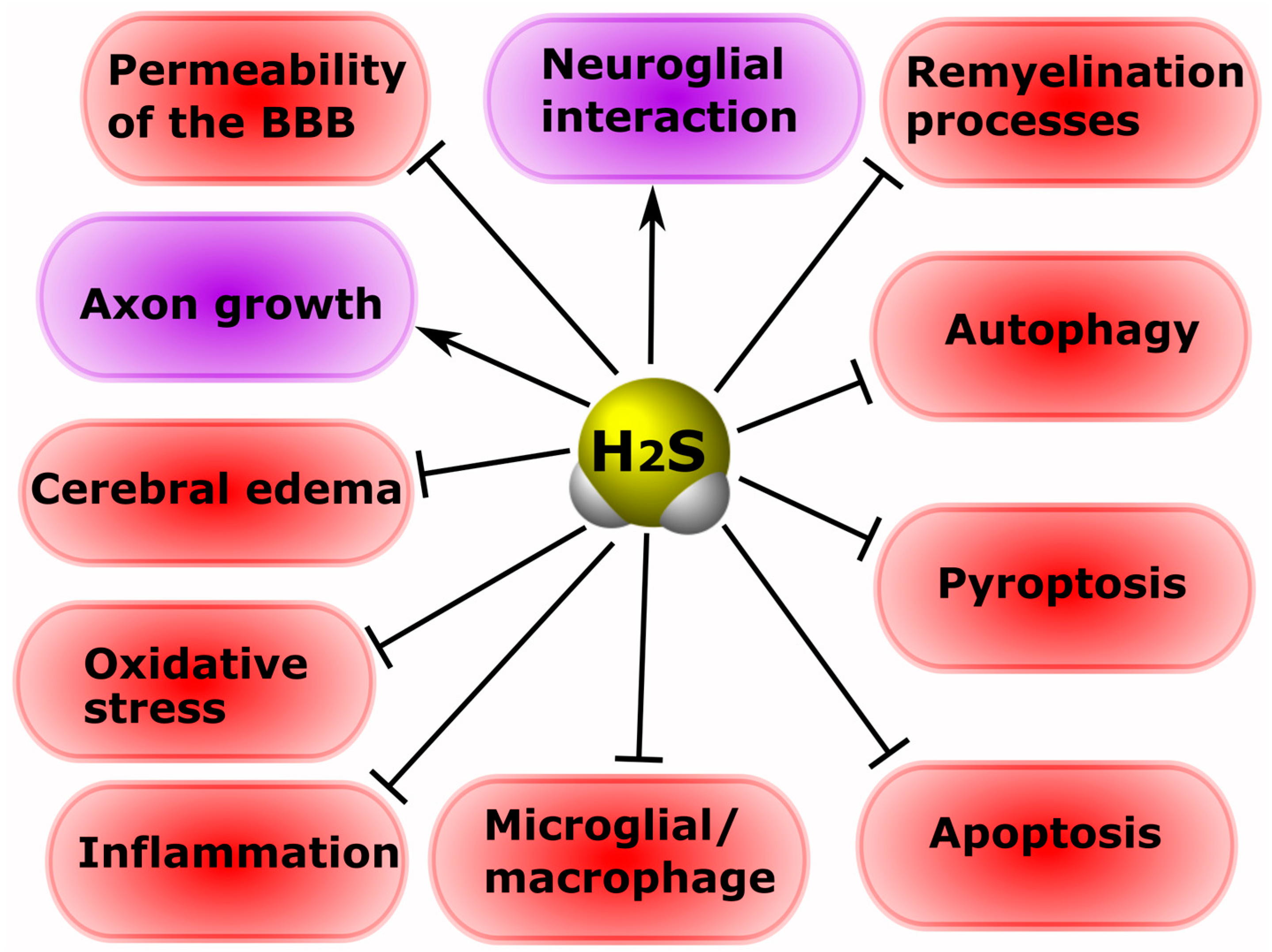
5. The Role of H2S in Cell Death in Neurotrauma
5.1. Participation of H2S in Oxidative Stress
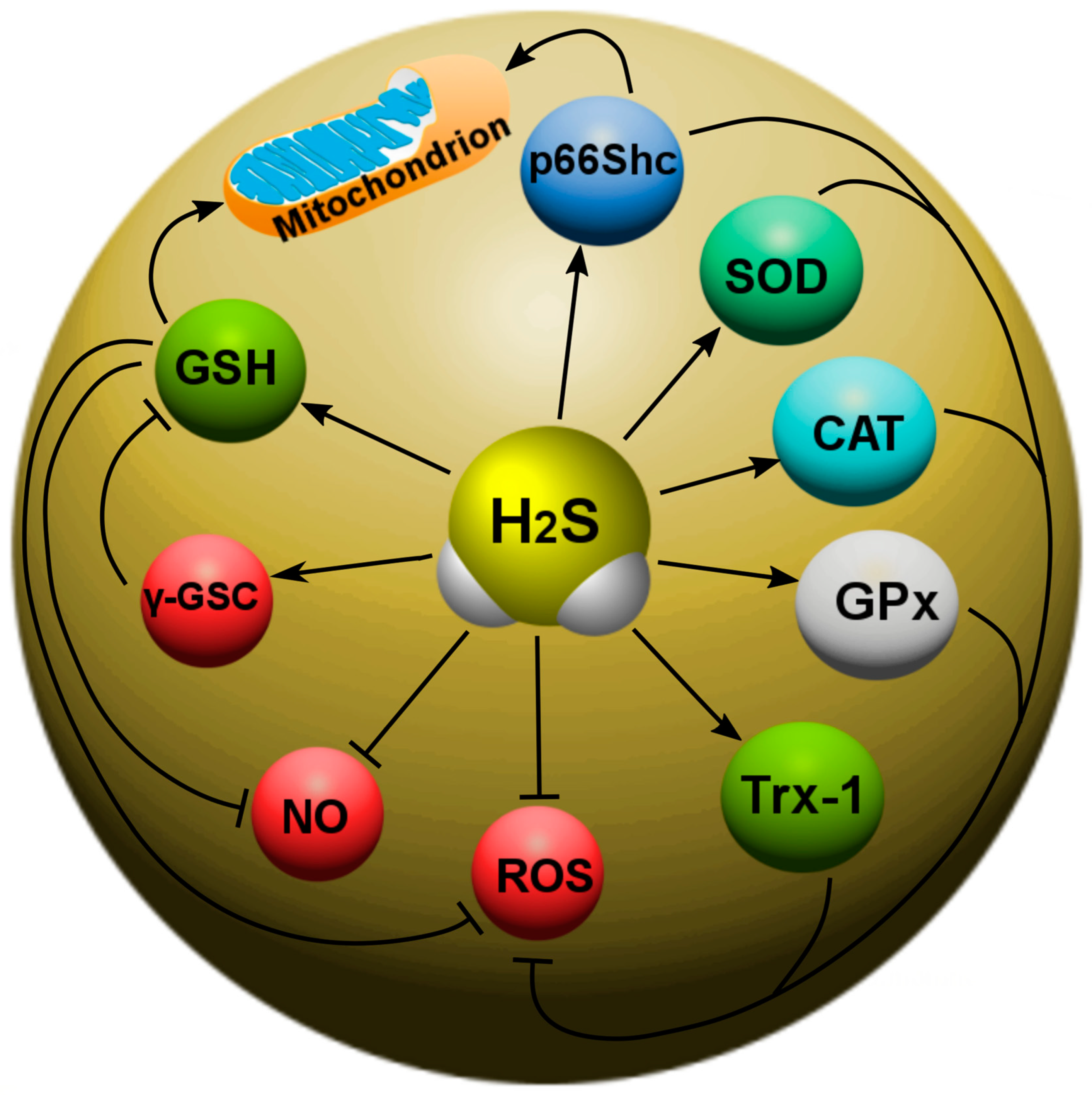
Recently, the main H2S-dependent biological effects in various neurotraumas are considered in the context of the regulation of oxidative stress. It is known that, at 37 °C and a pH of 7.4, more than 80% of H2S molecules dissolve in surface waters and dissociate into the ions H+, HS- and S2−. HS− is a powerful one-electron chemical reagent that effectively traps reactive oxygen species (ROS). Hydrosulfide anions are able to quench ROS by transferring a hydrogen atom or a single electron. The rate of this reaction is directly limited by diffusion. In this case, the reaction of hydrosulfide anions with molecular oxygen proceeds faster in the presence of divalent metal ions. H2S effectively interacts with hypochlorous acid (HClO), hydrogen peroxide (H2O2), lipid hydroperoxides and peroxynitrite (ONOO−), neutralizing their oxidative potential [91]. In addition, H2S itself is a reducing agent that can directly react and extinguish the superoxide anion (O2−), NO and its free radical products, as well as other ROS (Figure 5).
5.2. Modulation of the H2S Activity of NMDARs and Intracellular Ca2+ Homeostasis
5.3. Anti- and Pro-Inflammatory Effects of H2S
5.4. The Effect of H2S on the Level of Neurotrophic Factors
It is known that H2S is able to modulate the level of neurotrophic factors in normal and pathological conditions [26][105][106]. Thus, in a mouse TBI model, the administration of an H2S donor restored GDNF and NGF levels in damaged neural tissue, preserving their neuroprotective effects [64]. The use of a mitochondria-targeted H2S donor in middle cerebral artery occlusion has been reported to increase BDNF and NGF expression, reducing ischemic neuronal damage [26]. The administration of NaHS, a donor of H2S, increased BDNF levels, probably through activation of the transcription factor cAMP response element-binding protein (CREB), which regulates the gene for this neurotrophic factor in brain damage [107].
5.5. Effects of H2S on the Blood-Brain Barrier and Cerebral Edema
5.6. The Role of H2S in Remyelination Processes
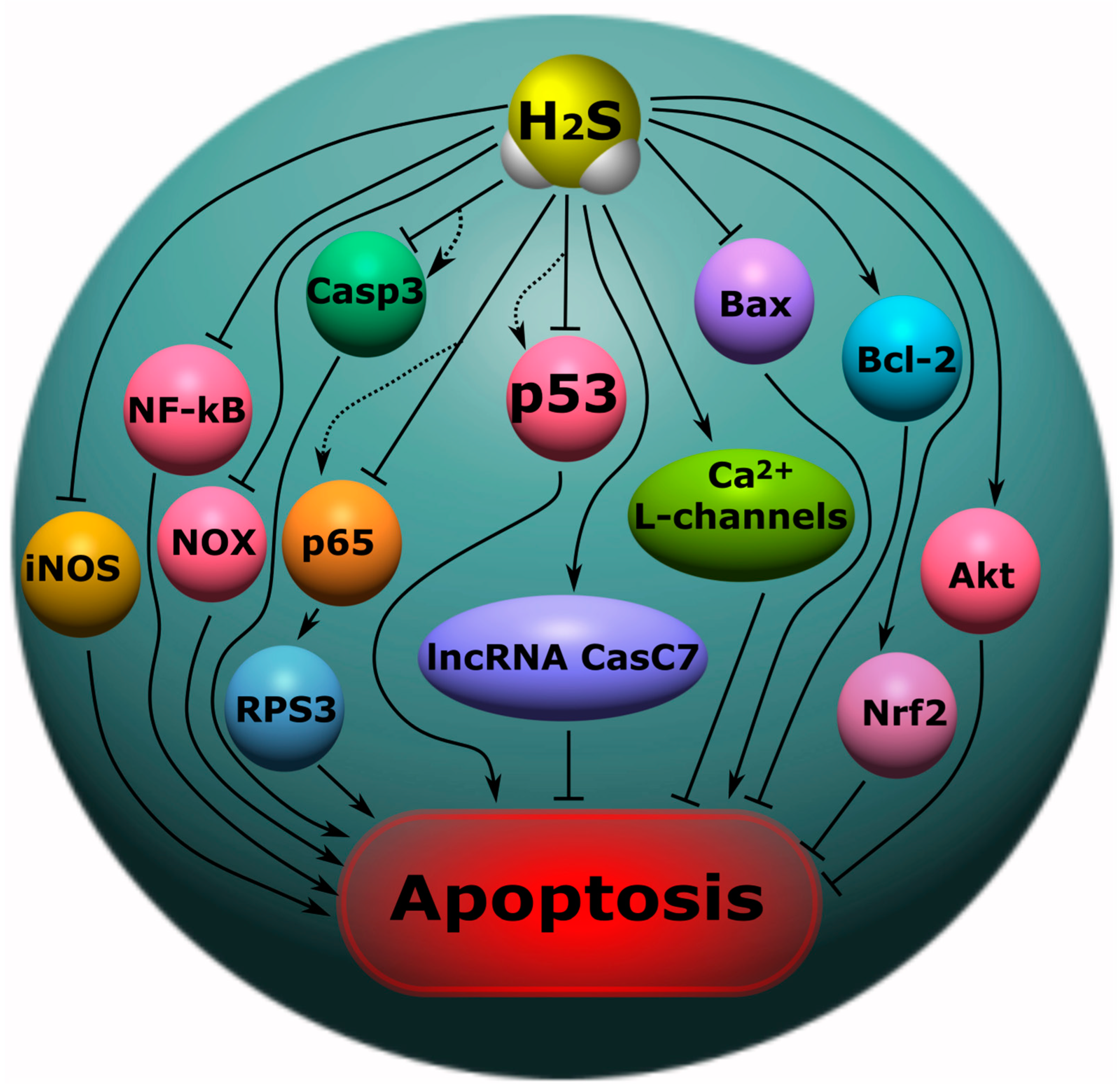
5.7. H2S-Associated Anti- and Pro-Apoptotic Signaling Mechanisms
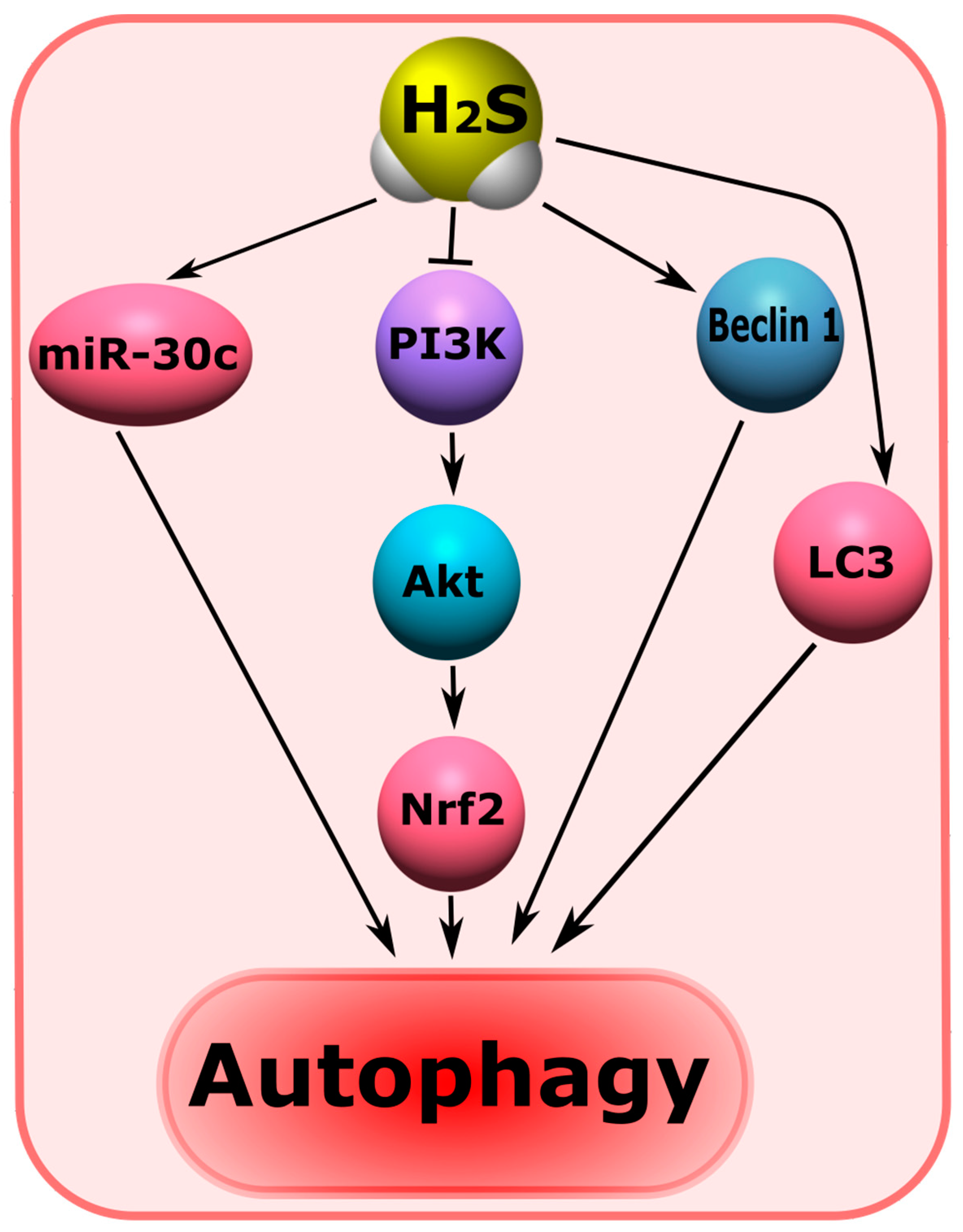
5.8. H2S-Associated Mechanisms of Autophagy
5.9. H2S-Associated Mechanisms of Ferroptosis
5.10. H2S-Associated Mechanisms of Pyroptosis
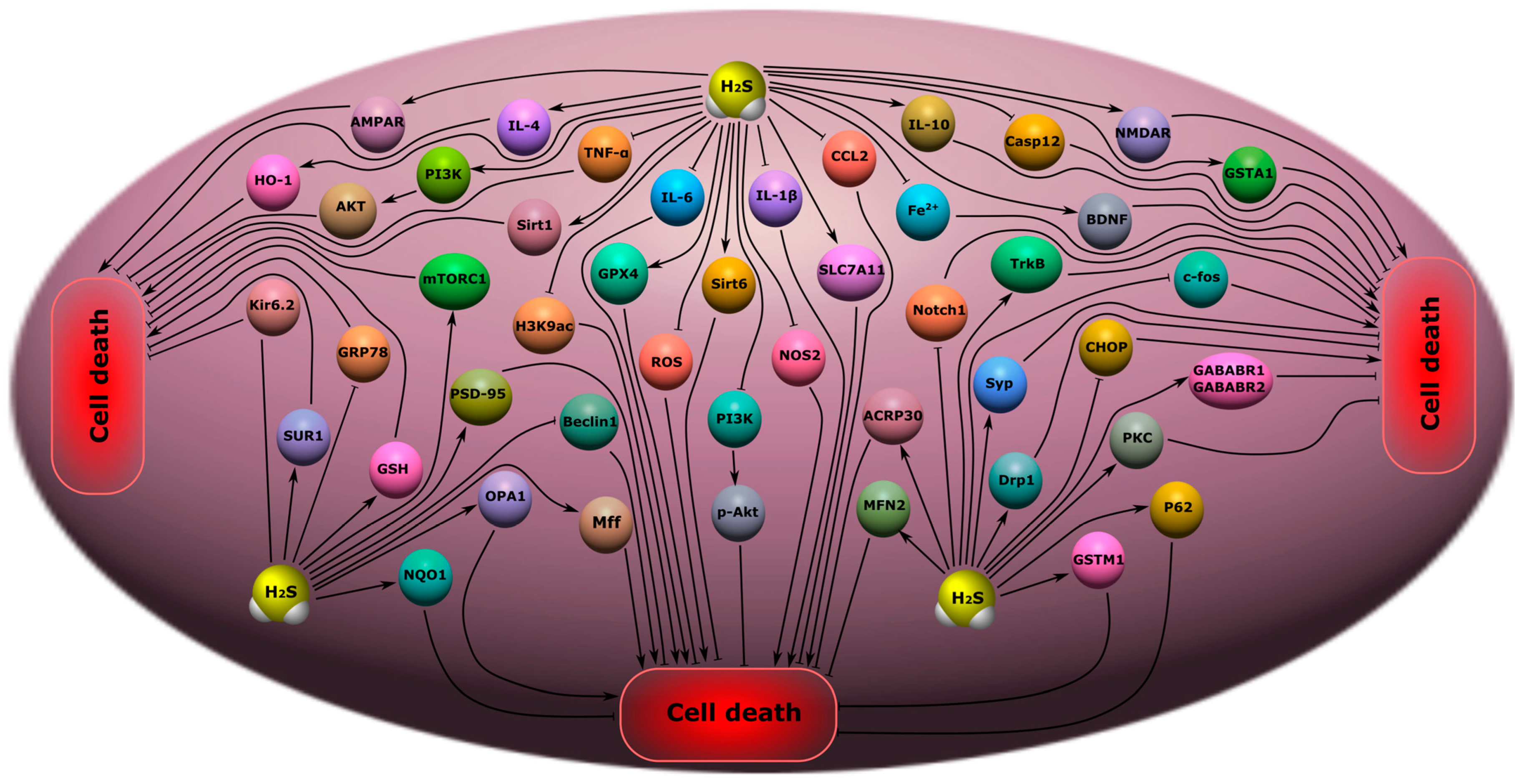
6. The Role of H2S in Mental Disorders and Neurodegenerative Diseases
6.1. Cognitive Impairment
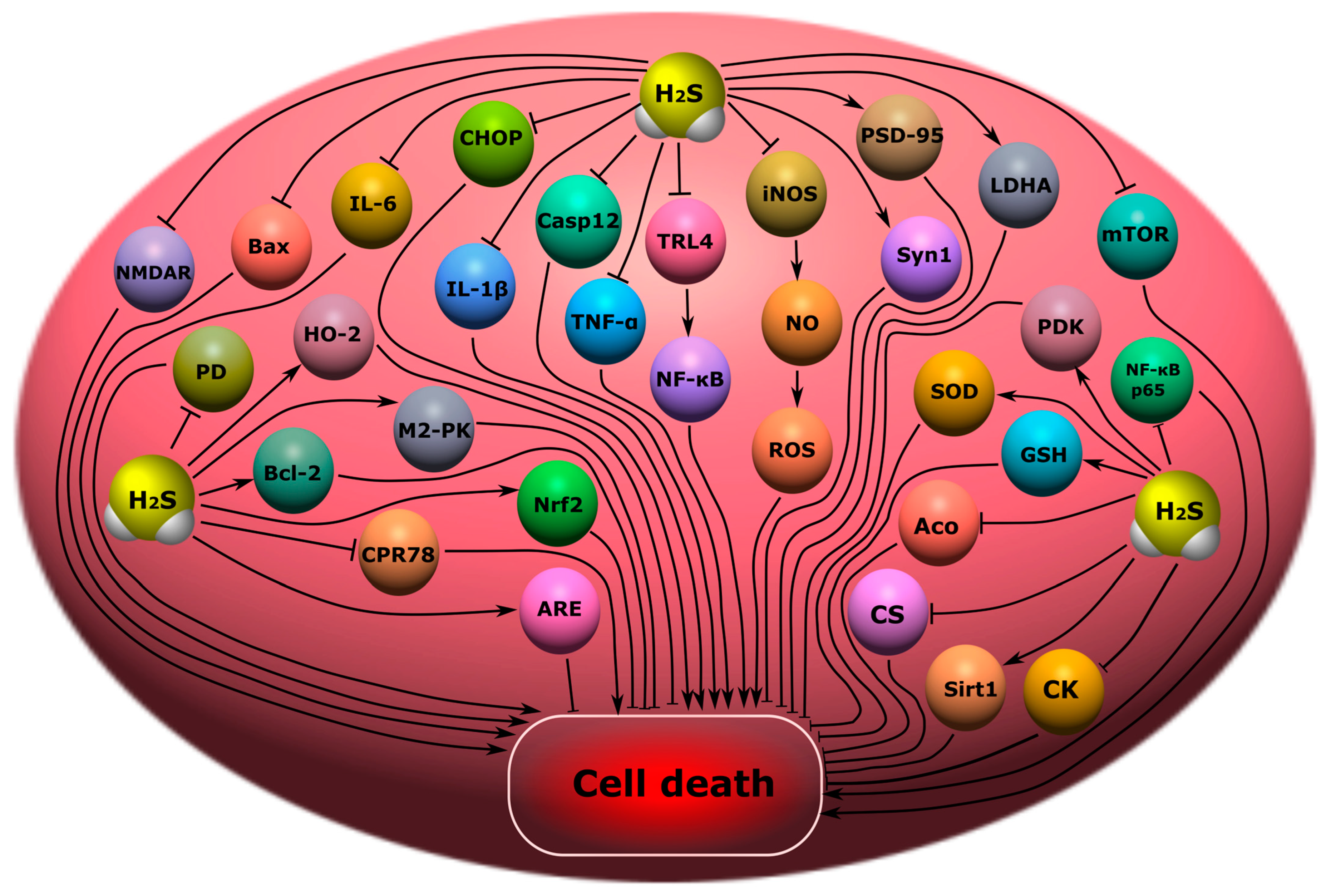
6.2. Encephalopathy
6.3. Depression and Anxiety Disorders
6.4. Epilepsy
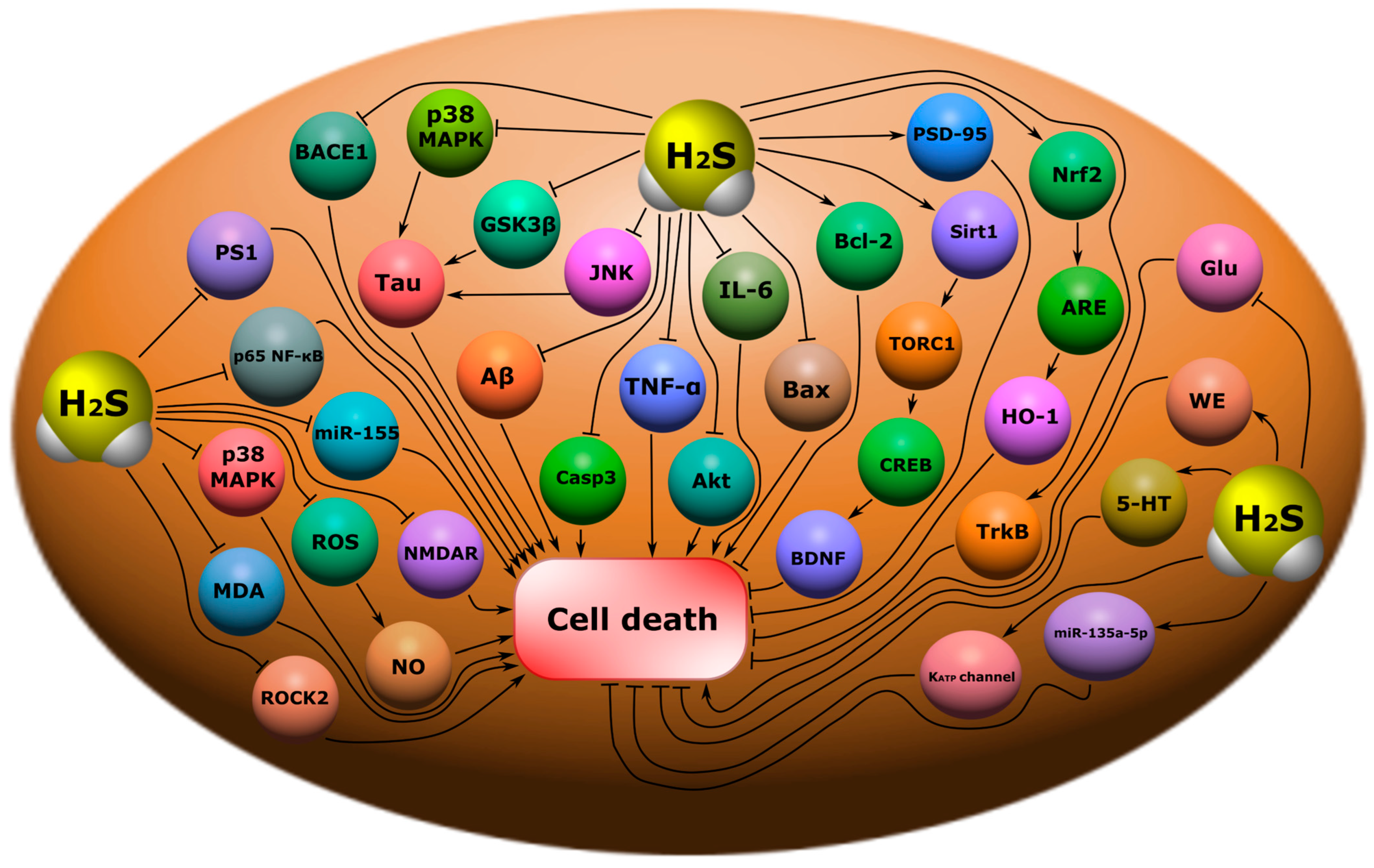
7. Neurodegenerative Diseases
7.1. Alzheimer’s Disease
7.2. Parkinson’s Disease
8. Therapeutic Approaches Using H2S as a Neuroprotector
9. Conclusions
References
- Furlan, J.C.; Gulasingam, S.; Craven, B.C. Epidemiology of War-Related Spinal Cord Injury Among Combatants: A Systematic Review. Glob. Spine J. 2019, 9, 545–558.
- Laskowitz, D.; Grant, G.E. Translational Research in Traumatic Brain Injury; CRC Press: Boca Raton, FL, USA, 2016.
- Rodkin, S.V.; Dzreyan, V.A.; Demyanenko, S.V.; Uzdensky, A.B. The Role of p53-Dependent Signaling Pathways in Survival and Death of Neurons and Glial Cells after Peripheral Nerve Injury. Biochem. (Moscow) Suppl. Ser. A Membr. Cell Biol. 2021, 15, 334–347.
- Karimi, S.A.; Hosseinmardi, N.; Janahmadi, M.; Sayyah, M.; Hajisoltani, R. The protective effect of hydrogen sulfide (H2S) on traumatic brain injury (TBI) induced memory deficits in rats. Brain Res. Bull. 2017, 134, 177–182.
- Calvillo, M.; Irimia, A. Neuroimaging and Psychometric Assessment of Mild Cognitive Impairment After Traumatic Brain Injury. Front. Psychol. 2020, 11, 1423.
- Sachdeva, R.; Nightingale, T.E.; Krassioukov, A.V. The Blood Pressure Pendulum following Spinal Cord Injury: Implications for Vascular Cognitive Impairment. Int. J. Mol. Sci. 2019, 20, 2464.
- Nightingale, T.E.; Zheng, M.M.Z.; Sachdeva, R.; Phillips, A.A.; Krassioukov, A.V. Diverse cognitive impairment after spinal cord injury is associated with orthostatic hypotension symptom burden. Physiol. Behav. 2020, 213, 112742.
- Chiaravalloti, N.D.; Weber, E.; Wylie, G.; Dyson-Hudson, T.; Wecht, J.M. Patterns of cognitive deficits in persons with spinal cord injury as compared with both age-matched and older individuals without spinal cord injury. J. Spinal Cord Med. 2020, 43, 88–97.
- Gupta, R.; Sen, N. Traumatic brain injury: A risk factor for neurodegenerative diseases. Rev. Neurosci. 2016, 27, 93–100.
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol. Psychiatry 2022, 91, 498–507.
- Yeh, T.-S.; Huang, Y.-P.; Wang, H.-I.; Pan, S.-L. Spinal cord injury and Parkinson’s disease: A population-based, propensity score-matched, longitudinal follow-up study. Spinal Cord 2016, 54, 1215–1219.
- Xu, X.-J.; Yang, M.-S.; Zhang, B.; Niu, F.; Dong, J.-Q.; Liu, B.-Y. Glucose metabolism: A link between traumatic brain injury and Alzheimer’s disease. Chin. J. Traumatol. 2021, 24, 5–10.
- White, D.L.; Kunik, M.E.; Yu, H.; Lin, H.L.; Richardson, P.A.; Moore, S.; Sarwar, A.I.; Marsh, L.; Jorge, R.E. Post-Traumatic Stress Disorder is Associated with further Increased Parkinson’s Disease Risk in Veterans with Traumatic Brain Injury. Ann. Neurol. 2020, 88, 33–41.
- Fox, G.B.; Fan, L.; Levasseur, R.A.; Faden, A.I. Sustained Sensory/Motor and Cognitive Deficits With Neuronal Apoptosis Following Controlled Cortical Impact Brain Injury in the Mouse. J. Neurotrauma 1998, 15, 599–614.
- Li, X.-N.; Chen, L.; Luo, B.; Li, X.; Wang, C.-Y.; Zou, W.; Zhang, P.; You, Y.; Tang, X.-Q. Hydrogen sulfide attenuates chronic restrain stress-induced cognitive impairment by upreglulation of Sirt1 in hippocampus. Oncotarget 2017, 8, 100396–100410.
- Liu, A.-H.; Chu, M.; Wang, Y.-P. Up-Regulation of Trem2 Inhibits Hippocampal Neuronal Apoptosis and Alleviates Oxidative Stress in Epilepsy via the PI3K/Akt Pathway in Mice. Neurosci. Bull. 2019, 35, 471–485.
- Saleem, S. Apoptosis, Autophagy, Necrosis and Their Multi Galore Crosstalk in Neurodegeneration. Neuroscience 2021, 469, 162–174.
- Ozdamar Unal, G.; Demirdas, A.; Nazıroglu, M.; Ovey, I.S. Agomelatine attenuates calcium signaling and apoptosis via the inhibition of TRPV1 channel in the hippocampal neurons of rats with chronic mild stress depression model. Behav. Brain Res. 2022, 434, 114033.
- Zhang, H.; Li, N.; Li, Z.; Li, Y.; Yu, Y.; Zhang, L. The Involvement of Caspases in Neuroinflammation and Neuronal Apoptosis in Chronic Pain and Potential Therapeutic Targets. Front. Pharmacol. 2022, 13, 1561.
- Mumtaz, S.; Rana, J.N.; Choi, E.H.; Han, I. Microwave Radiation and the Brain: Mechanisms, Current Status, and Future Prospects. Int. J. Mol. Sci. 2022, 23, 9288.
- Pérez-González, A.; Castañeda-Arriaga, R.; Guzmán-López, E.G.; Hernández-Ayala, L.F.; Galano, A. Chalcone Derivatives with a High Potential as Multifunctional Antioxidant Neuroprotectors. ACS Omega 2022, 7, 38254–38268.
- Ali, A.; Wang, Y.; Wu, L.; Yang, G. Gasotransmitter signaling in energy homeostasis and metabolic disorders. Free Radic. Res. 2021, 55, 83–105.
- Hendriks, K.D.; Maassen, H.; van Dijk, P.R.; Henning, R.H.; van Goor, H.; Hillebrands, J.-L. Gasotransmitters in health and disease: A mitochondria-centered view. Curr. Opin. Pharmacol. 2019, 45, 87–93.
- Sen, N. Functional and Molecular Insights of Hydrogen Sulfide Signaling and Protein Sulfhydration. J. Mol. Biol. 2017, 429, 543–561.
- Zhang, J.; Zhang, S.; Shan, H.; Zhang, M. Biologic Effect of Hydrogen Sulfide and Its Role in Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2020, 2020, 7301615.
- Pomierny, B.; Krzyżanowska, W.; Jurczyk, J.; Skórkowska, A.; Strach, B.; Szafarz, M.; Przejczowska-Pomierny, K.; Torregrossa, R.; Whiteman, M.; Marcinkowska, M.; et al. The Slow-Releasing and Mitochondria-Targeted Hydrogen Sulfide (H2S) Delivery Molecule AP39 Induces Brain Tolerance to Ischemia. Int. J. Mol. Sci. 2021, 22, 7816.
- Lv, S.; Wu, N.; Wang, Q.; Yang, L. Endogenous hydrogen sulfide alleviates methotrexate-induced cognitive impairment by attenuating endoplasmic reticulum stress-induced apoptosis via CHOP and caspase-12. Fundam. Clin. Pharmacol. 2020, 34, 559–570.
- Eto, K.; Asada, T.; Arima, K.; Makifuchi, T.; Kimura, H. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 293, 1485–1488.
- Sun, J.; Li, X.; Gu, X.; Du, H.; Zhang, G.; Wu, J.; Wang, F. Neuroprotective effect of hydrogen sulfide against glutamate-induced oxidative stress is mediated via the p53/glutaminase 2 pathway after traumatic brain injury. Aging 2021, 13, 7180–7189.
- Luo, Y.; Yang, X.; Zhao, S.; Wei, C.; Yin, Y.; Liu, T.; Jiang, S.; Xie, J.; Wan, X.; Mao, M.; et al. Hydrogen sulfide prevents OGD/R-induced apoptosis via improving mitochondrial dysfunction and suppressing an ROS-mediated caspase-3 pathway in cortical neurons. Neurochem. Int. 2013, 63, 826–831.
- Lu, D.; Wang, L.; Liu, G.; Wang, S.; Wang, Y.; Wu, Y.; Wang, J.; Sun, X. Role of hydrogen sulfide in subarachnoid hemorrhage. CNS Neurosci. Ther. 2022, 28, 805–817.
- Xie, Z.-Z.; Liu, Y.; Bian, J.-S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell. Longev. 2016, 2016, 6043038.
- Majid, A.S.A.; Majid, A.M.S.A.; Yin, Z.Q.; Ji, D. Slow Regulated Release of H2S Inhibits Oxidative Stress Induced Cell Death by Influencing Certain Key Signaling Molecules. Neurochem. Res. 2013, 38, 1375–1393.
- Tyagi, N.; Moshal, K.S.; Sen, U.; Vacek, T.P.; Kumar, M.; Hughes, W.M.; Kundu, S.; Tyagi, S.C. H2S Protects Against Methionine–Induced Oxidative Stress in Brain Endothelial Cells. Antioxid. Redox Signal. 2009, 11, 25–33.
- Xie, L.; Yu, S.; Yang, K.; Li, C.; Liang, Y. Hydrogen Sulfide Inhibits Autophagic Neuronal Cell Death by Reducing Oxidative Stress in Spinal Cord Ischemia Reperfusion Injury. Oxid. Med. Cell. Longev. 2017, 2017, 8640284.
- Deng, G.; Muqadas, M.; Adlat, S.; Zheng, H.; Li, G.; Zhu, P.; Nasser, M.I. Protective Effect of Hydrogen Sulfide on Cerebral Ischemia–Reperfusion Injury. Cell. Mol. Neurobiol. 2023, 43, 15–25.
- Bhatia, M. Role of Hydrogen Sulfide in the Pathology of Inflammation. Scientifica 2012, 2012, 159680.
- Ji, Y.; Li, Y.; Zhao, Z.; Li, P.; Xie, Y. Hydrogen Sulfide Overproduction Is Involved in Acute Ischemic Cerebral Injury Under Hyperhomocysteinemia. Front. Neurosci. 2020, 14, 582851.
- Calenic, B.; Yaegaki, K.; Ishkitiev, N.; Kumazawa, Y.; Imai, T.; Tanaka, T. p53-Pathway activity and apoptosis in hydrogen sulfide-exposed stem cells separated from human gingival epithelium. J. Periodontal Res. 2013, 48, 322–330.
- Calenic, B.; Yaegaki, K.; Kozhuharova, A.; Imai, T. Oral Malodorous Compound Causes Oxidative Stress and p53-Mediated Programmed Cell Death in Keratinocyte Stem Cells. J. Periodontol. 2010, 81, 1317–1323.
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3β and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118.
- Smith, C. Neurotrauma. Handb. Clin. Neurol. 2018, 145, 115–132.
- Chang, W.-T.W.; Badjatia, N. Neurotrauma. Emerg. Med. Clin. N. Am. 2014, 32, 889–905.
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord.–Drug Targets 2018, 17, 689–695.
- Sussman, E.S.; Pendharkar, A.V.; Ho, A.L.; Ghajar, J. Mild Traumatic Brain Injury and Concussion: Terminology and Classification; Elsevier: Amsterdam, The Netherlands, 2018; pp. 21–24.
- Robinson, C.P. Moderate and Severe Traumatic Brain Injury. Contin. Lifelong Learn. Neurol. 2021, 27, 1278–1300.
- Zhang, M.; Shan, H.; Chang, P.; Wang, T.; Dong, W.; Chen, X.; Tao, L. Hydrogen Sulfide Offers Neuroprotection on Traumatic Brain Injury in Parallel with Reduced Apoptosis and Autophagy in Mice. PLoS ONE 2014, 9, e87241.
- Wang, K.; Liu, B.; Ma, J. Research progress in traumatic brain penumbra. Chin. Med. J. 2014, 127, 1964–1968.
- Sun, G.; Gao, F.; Zhao, Z.; Sun, H.; Xu, W.; Wu, L.; He, Y. Endoplasmic reticulum stress-induced apoptosis in the penumbra aggravates secondary damage in rats with traumatic brain injury. Neural Regen. Res. 2016, 11, 1260–1266.
- Killen, M.J.; Giorgi-Coll, S.; Helmy, A.; Hutchinson, P.J.; Carpenter, K.L. Metabolism and inflammation: Implications for traumatic brain injury therapeutics. Expert Rev. Neurother. 2019, 19, 227–242.
- Zong, P.; Feng, J.; Yue, Z.; Li, Y.; Wu, G.; Sun, B.; He, Y.; Miller, B.; Yu, A.S.; Su, Z.; et al. Functional coupling of TRPM2 and extrasynaptic NMDARs exacerbates excitotoxicity in ischemic brain injury. Neuron 2022, 110, 1944–1958.e8.
- Moojen, V.K.M.; Damiani-Neves, M.; Bavaresco, D.V.; Pescador, B.B.; Comim, C.M.; Quevedo, J.; Boeck, C.R. NMDA preconditioning prevents object recognition memory impairment and increases brain viability in mice exposed to traumatic brain injury. Brain Res. 2012, 1466, 82–90.
- Zhou, K.; Sansur, C.; Xu, H.; Jia, X. The Temporal Pattern, Flux, and Function of Autophagy in Spinal Cord Injury. Int. J. Mol. Sci. 2017, 18, 466.
- Wang, H.; Wu, Y.; Han, W.; Li, J.; Xu, K.; Li, Z.; Wang, Q.; Xu, K.; Liu, Y.; Xie, L.; et al. Hydrogen Sulfide Ameliorates Blood-Spinal Cord Barrier Disruption and Improves Functional Recovery by Inhibiting Endoplasmic Reticulum Stress-Dependent Autophagy. Front. Pharmacol. 2018, 9, 858.
- Eckert, M.J.; Martin, M.J. Trauma. Surg. Clin. N. Am. 2017, 97, 1031–1045.
- Anjum, A.; Yazid, M.D.; Fauzi Daud, M.; Idris, J.; Ng, A.M.H.; Selvi Naicker, A.; Ismail, O.H.R.; Athi Kumar, R.K.; Lokanathan, Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. Int. J. Mol. Sci. 2020, 21, 7533.
- Goulart, C.; Martinez, A.B. Tubular conduits, cell-based therapy and exercise to improve peripheral nerve regeneration. Neural Regen. Res. 2015, 10, 565–567.
- Bhandari, P.S. Management of peripheral nerve injury. J. Clin. Orthop. Trauma 2019, 10, 862–866.
- Renthal, W.; Tochitsky, I.; Yang, L.; Cheng, Y.-C.; Li, E.; Kawaguchi, R.; Geschwind, D.H.; Woolf, C.J. Transcriptional Reprogramming of Distinct Peripheral Sensory Neuron Subtypes after Axonal Injury. Neuron 2020, 108, 128–144.e9.
- Hart, A.M.; Terenghi, G.; Wiberg, M. Neuronal death after peripheral nerve injury and experimental strategies for neuroprotection. Neurol. Res. 2008, 30, 999–1011.
- Navarro, X.; Vivó, M.; Valero-Cabré, A. Neural plasticity after peripheral nerve injury and regeneration. Prog. Neurobiol. 2007, 82, 163–201.
- Rishal, I.; Fainzilber, M. Axon–soma communication in neuronal injury. Nat. Rev. Neurosci. 2014, 15, 32–42.
- Patodia, S.; Raivich, G. Role of Transcription Factors in Peripheral Nerve Regeneration. Front. Mol. Neurosci. 2012, 5, 8.
- Campolo, M.; Esposito, E.; Ahmad, A.; Di Paola, R.; Paterniti, I.; Cordaro, M.; Bruschetta, G.; Wallace, J.L.; Cuzzocrea, S. Hydrogen sulfide-releasing cyclooxygenase inhibitor ATB-346 enhances motor function and reduces cortical lesion volume following traumatic brain injury in mice. J. Neuroinflamm. 2014, 11, 196.
- Oh, G.-S.; Pae, H.-O.; Lee, B.-S.; Kim, B.-N.; Kim, J.-M.; Kim, H.-R.; Jeon, S.B.; Jeon, W.K.; Chae, H.-J.; Chung, H.-T. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-κB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic. Biol. Med. 2006, 41, 106–119.
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129.
- Xiao, Q.; Ying, J.; Xiang, L.; Zhang, C. The biologic effect of hydrogen sulfide and its function in various diseases. Medicine 2018, 97, e13065.
- Khattak, S.; Rauf, M.A.; Khan, N.H.; Zhang, Q.-Q.; Chen, H.-J.; Muhammad, P.; Ansari, M.A.; Alomary, M.N.; Jahangir, M.; Zhang, C.-Y.; et al. Hydrogen Sulfide Biology and Its Role in Cancer. Molecules 2022, 27, 3389.
- Zhu, H.; Blake, S.; Chan, K.T.; Pearson, R.B.; Kang, J. Cystathionine β-Synthase in Physiology and Cancer. Biomed. Res. Int. 2018, 2018, 3205125.
- Lu, Z.; Zhao, T.; Tao, L.; Yu, Q.; Yang, Y.; Cheng, J.; Lu, S.; Ding, Q. Cystathionine β-Synthase-Derived Hydrogen Sulfide Correlates with Successful Aging in Mice. Rejuvenation Res. 2019, 22, 513–520.
- Kabil, O.; Banerjee, R. Enzymology of H2S Biogenesis, Decay and Signaling. Antioxid. Redox Signal. 2014, 20, 770–782.
- Zhang, M.Y.; Dugbartey, G.J.; Juriasingani, S.; Sener, A. Hydrogen Sulfide Metabolite, Sodium Thiosulfate: Clinical Applications and Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 6452.
- Park, B.S.; Kim, H.-W.; Rhyu, I.J.; Park, C.; Yeo, S.G.; Huh, Y.; Jeong, N.Y.; Jung, J. Hydrogen sulfide is essential for Schwann cell responses to peripheral nerve injury. J. Neurochem. 2015, 132, 230–242.
- Lupoli, R.; Di Minno, A.; Spadarella, G.; Franchini, M.; Sorrentino, R.; Cirino, G.; Di Minno, G. Methylation Reactions, the Redox Balance and Atherothrombosis: The Search for a Link with Hydrogen Sulfide. Semin. Thromb. Hemost. 2015, 41, 423–432.
- Lajin, B.; Francesconi, K.A. The hydrogen sulfide metabolite trimethylsulfonium is found in human urine. Sci. Rep. 2016, 6, 27038.
- Insko, M.A.; Deckwerth, T.L.; Hill, P.; Toombs, C.F.; Szabo, C. Detection of exhaled hydrogen sulphide gas in rats exposed to intravenous sodium sulphide. Br. J. Pharmacol. 2009, 157, 944–951.
- Wang, R. Hydrogen Sulfide: The Third Gasotransmitter in Biology and Medicine. Antioxid. Redox Signal. 2010, 12, 1061–1064.
- Yakovlev, A.V.; Kurmasheva, E.D.; Ishchenko, Y.; Giniatullin, R.; Sitdikova, G.F. Age-Dependent, Subunit Specific Action of Hydrogen Sulfide on GluN1/2A and GluN1/2B NMDA Receptors. Front. Cell. Neurosci. 2017, 11, 375.
- Munaron, L.; Avanzato, D.; Moccia, F.; Mancardi, D. Hydrogen sulfide as a regulator of calcium channels. Cell Calcium 2013, 53, 77–84.
- Disbrow, E.; Stokes, K.Y.; Ledbetter, C.; Patterson, J.; Kelley, R.; Pardue, S.; Reekes, T.; Larmeu, L.; Batra, V.; Yuan, S.; et al. Plasma hydrogen sulfide: A biomarker of Alzheimer’s disease and related dementias. Alzheimer’s Dement. 2021, 17, 1391–1402.
- Liu, L.; Wang, J.; Wang, H. Hydrogen sulfide alleviates oxidative stress injury and reduces apoptosis induced by MPP+ in Parkinson’s disease cell model. Mol. Cell. Biochem. 2020, 472, 231–240.
- Zhang, M.; Shan, H.; Wang, T.; Liu, W.; Wang, Y.; Wang, L.; Zhang, L.; Chang, P.; Dong, W.; Chen, X.; et al. Dynamic Change of Hydrogen Sulfide After Traumatic Brain Injury and its Effect in Mice. Neurochem. Res. 2013, 38, 714–725.
- Zhang, X.; Bian, J.-S. Hydrogen Sulfide: A Neuromodulator and Neuroprotectant in the Central Nervous System. ACS Chem. Neurosci. 2014, 5, 876–883.
- Jiang, X.; Huang, Y.; Lin, W.; Gao, D.; Fei, Z. Protective effects of hydrogen sulfide in a rat model of traumatic brain injury via activation of mitochondrial adenosine triphosphate–sensitive potassium channels and reduction of oxidative stress. J. Surg. Res. 2013, 184, e27–e35.
- Xu, K.; Wu, F.; Xu, K.; Li, Z.; Wei, X.; Lu, Q.; Jiang, T.; Wu, F.; Xu, X.; Xiao, J.; et al. NaHS restores mitochondrial function and inhibits autophagy by activating the PI3K/Akt/mTOR signalling pathway to improve functional recovery after traumatic brain injury. Chem. Biol. Interact. 2018, 286, 96–105.
- Wang, R.; Wu, X.-X.; Tian, Z.; Hu, T.; Cai, C.; Wu, G.-P.; Jiang, G.-B.; Liu, B. Sustained release of hydrogen sulfide from anisotropic ferrofluid hydrogel for the repair of spinal cord injury. Bioact. Mater. 2023, 23, 118–128.
- Chen, X.; Huang, X.; Liu, C.; Li, S.; Yang, Z.; Zhang, F.; Chen, X.; Shan, H.; Tao, L.; Zhang, M. Surface-fill H2S-releasing silk fibroin hydrogel for brain repair through the repression of neuronal pyroptosis. Acta Biomater. 2022, 154, 259–274.
- Liu, Y.; Pan, L.; Jiang, A.; Yin, M. Hydrogen sulfide upregulated lncRNA CasC7 to reduce neuronal cell apoptosis in spinal cord ischemia-reperfusion injury rat. Biomed. Pharmacother. 2018, 98, 856–862.
- Kida, K.; Marutani, E.; Nguyen, R.K.; Ichinose, F. Inhaled hydrogen sulfide prevents neuropathic pain after peripheral nerve injury in mice. Nitric Oxide 2015, 46, 87–92.
- Jung, J.; Jeong, N. Hydrogen sulfide controls peripheral nerve degeneration and regeneration: A novel therapeutic strategy for peripheral demyelinating disorders or nerve degenerative diseases. Neural Regen. Res. 2014, 9, 2119–2121.
- Predmore, B.L.; Lefer, D.J.; Gojon, G. Hydrogen Sulfide in Biochemistry and Medicine. Antioxid. Redox Signal. 2012, 17, 119–140.
- Bruce King, S. Potential biological chemistry of hydrogen sulfide (H2S) with the nitrogen oxides. Free Radic. Biol. Med. 2013, 55, 21–34.
- Searcy, D.G.; Whitehead, J.P.; Maroney, M.J. Interaction of Cu, Zn Superoxide Dismutase with Hydrogen Sulfide. Arch. Biochem. Biophys. 1995, 318, 251–263.
- Vega-Vela, N.E.; Osorio, D.; Avila-Rodriguez, M.; Gonzalez, J.; García-Segura, L.M.; Echeverria, V.; Barreto, G.E. L-Type Calcium Channels Modulation by Estradiol. Mol. Neurobiol. 2017, 54, 4996–5007.
- Jiang, M.C.; Birch, D.V.; Heckman, C.J.; Tysseling, V.M. The Involvement of CaV1.3 Channels in Prolonged Root Reflexes and Its Potential as a Therapeutic Target in Spinal Cord Injury. Front. Neural Circuits 2021, 15, 642111.
- Alles, S.R.; Garcia, E.; Balasubramanyan, S.; Jones, K.; Tyson, J.R.; Joy, T.; Snutch, T.P.; Smith, P.A. Peripheral nerve injury increases contribution of L-type calcium channels to synaptic transmission in spinal lamina II: Role of α2δ–1 subunits. Mol. Pain 2018, 14, 174480691876580.
- Ihbe, N.; Le Prieult, F.; Wang, Q.; Distler, U.; Sielaff, M.; Tenzer, S.; Thal, S.C.; Mittmann, T. Adaptive Mechanisms of Somatostatin-Positive Interneurons after Traumatic Brain Injury through a Switch of α Subunits in L-Type Voltage-Gated Calcium Channels. Cereb. Cortex 2022, 32, 1093–1109.
- Tang, G.; Wu, L.; Wang, R. Interaction of hydrogen sulfide with ion channels. Clin. Exp. Pharmacol. Physiol. 2010, 37, 753–763.
- Nagai, Y.; Tsugane, M.; Oka, J.; Kimura, H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004, 18, 557–559.
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153.
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258.
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209.
- Éva Sikura, K.; Combi, Z.; Potor, L.; Szerafin, T.; Hendrik, Z.; Méhes, G.; Gergely, P.; Whiteman, M.; Beke, L.; Fürtös, I.; et al. Hydrogen sulfide inhibits aortic valve calcification in heart via regulating RUNX2 by NF-κB, a link between inflammation and mineralization. J. Adv. Res. 2021, 27, 165–176.
- Rose, P.; Zhu, Y.-Z.; Moore, P.K. Hydrogen Sulfide and the Immune System. Adv. Exp. Med. Biol. 2021, 1315, 99–128.
- Li, X.; Zhuang, Y.-Y.; Wu, L.; Xie, M.; Gu, H.-F.; Wang, B.; Tang, X.-Q. Hydrogen Sulfide Ameliorates Cognitive Dysfunction in Formaldehyde-Exposed Rats: Involvement in the Upregulation of Brain-Derived Neurotrophic Factor. Neuropsychobiology 2020, 79, 119–130.
- Mohseni, F.; Bagheri, F.; Rafaiee, R.; Norozi, P.; Khaksari, M. Hydrogen sulfide improves spatial memory impairment via increases of BDNF expression and hippocampal neurogenesis following early postnatal alcohol exposure. Physiol. Behav. 2020, 215, 112784.
- Li, T.; Liu, H.; Xue, H.; Zhang, J.; Han, X.; Yan, S.; Bo, S.; Liu, S.; Yuan, L.; Deng, L.; et al. Neuroprotective Effects of Hydrogen Sulfide Against Early Brain Injury and Secondary Cognitive Deficits Following Subarachnoid Hemorrhage. Brain Pathol. 2017, 27, 51–63.
- Wang, Y.; Jia, J.; Ao, G.; Hu, L.; Liu, H.; Xiao, Y.; Du, H.; Alkayed, N.J.; Liu, C.-F.; Cheng, J. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J. Neurochem. 2014, 129, 827–838.
- Kumar, M.; Sandhir, R. Hydrogen sulfide attenuates hyperhomocysteinemia-induced blood-brain barrier permeability by inhibiting MMP-9. Int. J. Neurosci. 2022, 132, 1061–1071.
- Cai, S.; Li, Q.; Fan, J.; Zhong, H.; Cao, L.; Duan, M. Therapeutic Hypothermia Combined with Hydrogen Sulfide Treatment Attenuated Early Blood–Brain Barrier Disruption and Brain Edema Induced by Cardiac Arrest and Resuscitation in Rat Model. Neurochem. Res. 2022, 48, 967–979.
- Li, H.; Zhu, L.; Feng, J.; Hu, X.; Li, C.; Zhang, B. Hydrogen Sulfide Decreases Blood-Brain Barrier Damage via Regulating Protein Kinase C and Tight Junction After Cardiac Arrest in Rats. Cell. Physiol. Biochem. 2018, 47, 994–1006.
- Haber, M.; James, J.; Kim, J.; Sangobowale, M.; Irizarry, R.; Ho, J.; Nikulina, E.; Grin’kina, N.M.; Ramadani, A.; Hartman, I.; et al. Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes and modifies neuroinflammation in a rat model of mild traumatic brain injury. J. Cereb. Blood Flow Metab. 2018, 38, 1312–1326.
- Mekhail, M.; Almazan, G.; Tabrizian, M. Oligodendrocyte-protection and remyelination post-spinal cord injuries: A review. Prog. Neurobiol. 2012, 96, 322–339.
- Svennigsen, Å.; Dahlin, L. Repair of the Peripheral Nerve—Remyelination that Works. Brain Sci. 2013, 3, 1182–1197.
- Iqbal, I.K.; Bajeli, S.; Sahu, S.; Bhat, S.A.; Kumar, A. Hydrogen sulfide-induced GAPDH sulfhydration disrupts the CCAR2-SIRT1 interaction to initiate autophagy. Autophagy 2021, 17, 3511–3529.
- Fukuto, J.M.; Vega, V.S.; Works, C.; Lin, J. The chemical biology of hydrogen sulfide and related hydropersulfides: Interactions with biologically relevant metals and metalloproteins. Curr. Opin. Chem. Biol. 2020, 55, 52–58.
- Jiang, M.; Qi, L.; Li, L.; Li, Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 2020, 6, 112.
- Dzreyan, V.; Rodkin, S.; Nikul, V.; Pitinova, M.; Uzdensky, A. The Expression of E2F1, p53, and Caspase 3 in the Rat Dorsal Root Ganglia After Sciatic Nerve Transection. J. Mol. Neurosci. 2021, 71, 826–835.
- Xu, C.; Zhang, M.; Zhang, G.; Yan, S.; Yan, W. Hydrogen Sulfide Improves Functional Recovery in Rat Traumatic Spinal Cord Injury Model by Inducing Nuclear Translocation of NF-E2-Related Factor 2. Biol. Pharm. Bull. 2021, 44, b21-00259.
- Sarkar, C.; Zhao, Z.; Aungst, S.; Sabirzhanov, B.; Faden, A.I.; Lipinski, M.M. Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 2014, 10, 2208–2222.
- Zhang, J.; Shi, C.; Wang, H.; Gao, C.; Chang, P.; Chen, X.; Shan, H.; Zhang, M.; Tao, L. Hydrogen sulfide protects against cell damage through modulation of PI3K/Akt/Nrf2 signaling. Int. J. Biochem. Cell Biol. 2019, 117, 105636.
- Zhao, G.; Qi, L.; Wang, Y.; Li, X.; Li, Q.; Tang, X.; Wang, X.; Wu, C. Antagonizing effects of curcumin against mercury-induced autophagic death and trace elements disorder by regulating PI3K/AKT and Nrf2 pathway in the spleen. Ecotoxicol. Environ. Saf. 2021, 222, 112529.
- Luo, L.-F.; Qin, L.-Y.; Wang, J.-X.; Guan, P.; Wang, N.; Ji, E.-S. Astragaloside IV Attenuates the Myocardial Injury Caused by Adriamycin by Inhibiting Autophagy. Front. Pharmacol. 2021, 12, 669782.
- Jin, R.; Yang, R.; Cui, C.; Zhang, H.; Cai, J.; Geng, B.; Chen, Z. Ferroptosis due to Cystathionine γ Lyase/Hydrogen Sulfide Downregulation under High Hydrostatic Pressure Exacerbates VSMC Dysfunction. Front. Cell Dev. Biol. 2022, 10, 829316.
- Yu, M.; Wang, W.; Dang, J.; Liu, B.; Xu, J.; Li, J.; Liu, Y.; He, L.; Ying, Y.; Cai, J.; et al. Hydrogen sulfide protects retinal pigment epithelium cells against ferroptosis through the AMPK- and p62-dependent non-canonical NRF2-KEAP1 pathway. Exp. Cell Res. 2023, 422, 113436.
- Yu, Y.; Li, X.; Wu, X.; Li, X.; Wei, J.; Chen, X.; Sun, Z.; Zhang, Q. Sodium hydrosulfide inhibits hemin-induced ferroptosis and lipid peroxidation in BV2 cells via the CBS/H2S system. Cell. Signal. 2023, 104, 110594.
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128.
- Hu, X.; Chen, H.; Xu, H.; Wu, Y.; Wu, C.; Jia, C.; Li, Y.; Sheng, S.; Xu, C.; Xu, H.; et al. Role of Pyroptosis in Traumatic Brain and Spinal Cord Injuries. Int. J. Biol. Sci. 2020, 16, 2042–2050.
- Yang, K.-L.; Li, W.-H.; Liu, Y.-J.; Wei, Y.-J.; Ren, Y.-K.; Mai, C.-D.; Zhang, S.-Y.; Zuo, Y.; Sun, Z.-Z.; Li, D.-L.; et al. Hydrogen Sulfide Attenuates Neuroinflammation by Inhibiting the NLRP3/Caspase-1/GSDMD Pathway in Retina or Brain Neuron following Rat Ischemia/Reperfusion. Brain Sci. 2022, 12, 1245.
- Duan, H.; Li, L.; Shen, S.; Ma, Y.; Yin, X.; Liu, Z.; Yuan, C.; Wang, Y.; Zhang, J. Hydrogen Sulfide Reduces Cognitive Impairment in Rats After Subarachnoid Hemorrhage by Ameliorating Neuroinflammation Mediated by the TLR4/NF-κB Pathway in Microglia. Front. Cell. Neurosci. 2020, 14, 210.
- Chu, Q.-J.; He, L.; Zhang, W.; Liu, C.-L.; Ai, Y.-Q.; Zhang, Q. Hydrogen sulfide attenuates surgical trauma-induced inflammatory response and cognitive deficits in mice. J. Surg. Res. 2013, 183, 330–336.
- Jin, X.; Chen, D.; Wu, F.; Zhang, L.; Huang, Y.; Lin, Z.; Wang, X.; Wang, R.; Xu, L.; Chen, Y. Hydrogen Sulfide Protects Against Ammonia-Induced Neurotoxicity Through Activation of Nrf2/ARE Signaling in Astrocytic Model of Hepatic Encephalopathy. Front. Cell. Neurosci. 2020, 14, 573422.
- Chen, W.-L.; Xie, B.; Zhang, C.; Xu, K.-L.; Niu, Y.-Y.; Tang, X.-Q.; Zhang, P.; Zou, W.; Hu, B.; Tian, Y. Antidepressant-like and anxiolytic-like effects of hydrogen sulfide in behavioral models of depression and anxiety. Behav. Pharmacol. 2013, 24, 590–597.
- Kang, X.; Jiang, L.; Lan, F.; Tang, Y.-Y.; Zhang, P.; Zou, W.; Chen, Y.-J.; Tang, X.-Q. Hydrogen sulfide antagonizes sleep deprivation-induced depression- and anxiety-like behaviors by inhibiting neuroinflammation in a hippocampal Sirt1-dependent manner. Brain Res. Bull. 2021, 177, 194–202.
- Cho, C.; Zeigler, M.; Mizuno, S.; Morrison, R.S.; Totah, R.A.; Barker-Haliski, M. Reductions in Hydrogen Sulfide and Changes in Mitochondrial Quality Control Proteins Are Evident in the Early Phases of the Corneally Kindled Mouse Model of Epilepsy. Int. J. Mol. Sci. 2022, 23, 1434.
- Vandini, E.; Ottani, A.; Zaffe, D.; Calevro, A.; Canalini, F.; Cavallini, G.M.; Rossi, R.; Guarini, S.; Giuliani, D. Mechanisms of Hydrogen Sulfide against the Progression of Severe Alzheimer’s Disease in Transgenic Mice at Different Ages. Pharmacology 2019, 103, 50–60.
- Cao, X.; Cao, L.; Ding, L.; Bian, J. A New Hope for a Devastating Disease: Hydrogen Sulfide in Parkinson’s Disease. Mol. Neurobiol. 2017, 55, 3789–3799.
- Xue, X.; Bian, J.-S. Neuroprotective Effects of Hydrogen Sulfide in Parkinson’s Disease Animal Models. Methods Enzymol. 2015, 554, 169–186.
- Xie, L.; Hu, L.-F.; Teo, X.Q.; Tiong, C.X.; Tazzari, V.; Sparatore, A.; del Soldato, P.; Dawe, G.S.; Bian, J.-S. Therapeutic Effect of Hydrogen Sulfide-Releasing L-Dopa Derivative ACS84 on 6-OHDA-Induced Parkinson’s Disease Rat Model. PLoS ONE 2013, 8, e60200.
- Huang, Y.; Omorou, M.; Gao, M.; Mu, C.; Xu, W.; Xu, H. Hydrogen sulfide and its donors for the treatment of cerebral ischaemia-reperfusion injury: A comprehensive review. Biomed. Pharmacother. 2023, 161, 114506.
- Whiteman, M.; Perry, A.; Zhou, Z.; Bucci, M.; Papapetropoulos, A.; Cirino, G.; Wood, M.E. Phosphinodithioate and Phosphoramidodithioate Hydrogen Sulfide Donors. In Chemistry, Biochemistry and Pharmacology of Hydrogen Sulfide; Springer: Berlin/Heidelberg, Germany, 2015; pp. 337–363.
- Huang, C.W.; Feng, W.; Peh, M.T.; Peh, K.; Dymock, B.W.; Moore, P.K. A novel slow-releasing hydrogen sulfide donor, FW1256, exerts anti-inflammatory effects in mouse macrophages and in vivo. Pharmacol. Res. 2016, 113, 533–546.

