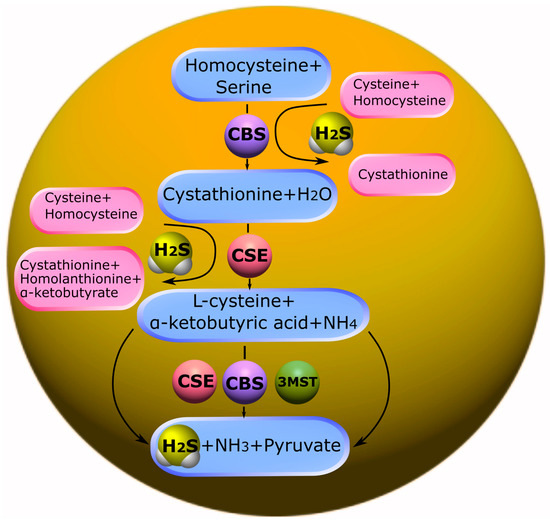
CBS is predominantly expressed in the brain, liver, kidneys, and pancreas. CBS catalyzes the condensation of homocysteine (Hcy) with serine to form cystathionine. Subsequently, cystathionine undergoes proteolysis by the enzyme CSE. This leads to the formation of cysteine, which is a precursor of glutathione. It should be noted that in addition to the canonical pathway, CBS is involved in desulfurization reactions that lead to the formation of endogenous H
2S. The formation of H
2S may be affected by a thiol-cysteine reaction catalyzed by CBS with the release of s-thiolate. Cysteine may also undergo hydrolysis by CSE to form H
2S, as well as pyruvate and ammonia. Disruption of the mechanisms of regulation of CBS is associated with a change in the levels of Hcy and/or H
2S and, inevitably, leads to various pathological conditions (
Figure 1) [
106,
107].
3.2. Catabolism of H2S
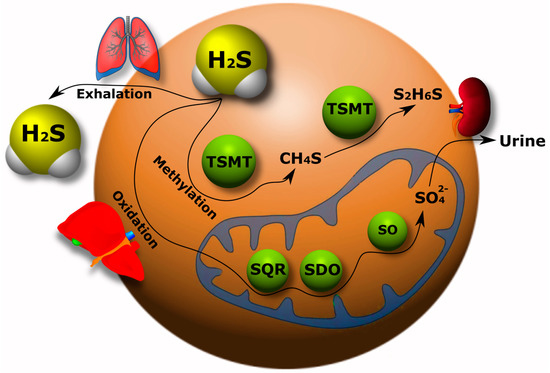
The catabolism of H
2S has been studied much less than its synthesis in the body. Currently, three pathways of H
2S catabolism are mainly known: oxidation, methylation and exhalation. The oxidation of H
2S mainly occurs in hepatocytes. Here, H
2S is oxidized by mitochondrial enzymes to sulfate with the intermediates persulfide (RSSH), sulfite (SO
32−), and thiosulfate (S
2O
32−) [
111,
112].
As a result, most of the H
2S is excreted in the urine in the form of sulfate. It has been shown that the increase in sulfide oxidation in the kidneys, heart and liver upon administration of exogenous H
2S is due to an increase in quinone oxidoreductase (SQR) (
Figure 2). However, this effect was not observed in the brain tissue, which indicates a defect in the oxidation of sulfides in the nervous tissue of the brain [
113].
Figure 2. H2S catabolism pathways in the body: oxidation, methylation and exhalation. TSMT, thiol-S-methyl transferase; SQR, quinone oxidoreductase; SDO, sulfur deoxygenase; SO, sulfite oxidase.
Free H
2S exists in low concentration in the blood and decays rapidly. Therefore, it will probably not all be transported to the liver for disposal. The question of H
2S catabolism in the brain via alternative pathways independent of the liver and kidneys remains open [
31].
The methylation of H
2S occurs primarily in the cytoplasm in contrast to the oxidative catabolic pathway of this gasotransmitter. First, H
2S is methylated to methane thiol, and then it is methylated to a non-toxic dimethyl sulfide by a thiol-S-methyl transferase (TSMT) (
Figure 2). The methylation of sulfides has been found to be a significantly slower process than oxidation [
114,
115]. H
2S can be excreted from the body through lung tissue (
Figure 2) [
31,
116].
3.3. Various Biological Effects of Endogenous H2S
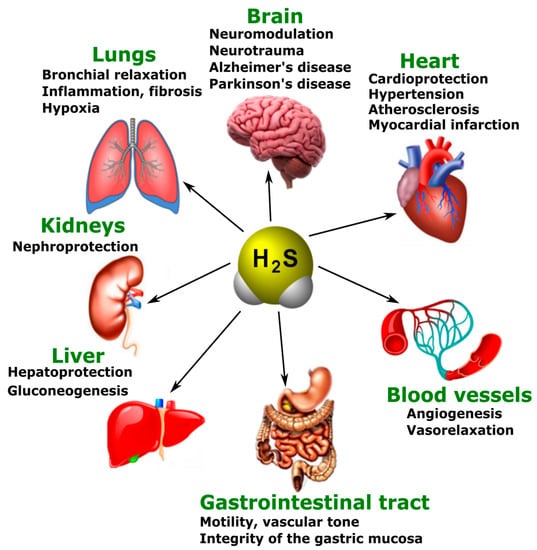
The main physiological effects of H
2S are neuromodulation, regulation of vascular tone and oxidative stress, anti-inflammatory action, angiogenesis, and energy generation. However, these effects do not exhaust the diversity of the biological actions of H
2S. Presently, the list of functions performed by this gaseous signaling agent is constantly expanding (
Figure 3) [
117].
Figure 3. The participation of H2S in normal and pathological conditions in the brain, heart, blood vessels, gastrointestinal tract, liver, kidneys, and lungs.
Just like NO, H
2S is a new generation neurotransmitter that has also been shown to have pronounced neuroprotective effects. In physiological conditions, H
2S has a role in learning and memory processes. It facilitates long-term potentiation in the hippocampus by activating NMDARs associated with Ca
2+ channels [
118,
119]. Disturbances in the endogenous production and metabolism of H
2S have been observed in neurodegenerative diseases, such as Alzheimer’s (AD) and Parkinson’s disease (PD) [
120,
121].
4. Endogenous and Exogenous H2S in Neurotrauma
4.1. Endogenous H2S Levels in Neurotrauma
Recently, several experiments have been developed to study and detect changes in the concentration of H
2S in neurotrauma in both animal and human models. The concentration of H
2S has been shown to be a dynamic system. The level of the endogenous expression of H
2S and CBS in the blood and brain tends to decrease after neurotrauma. Zhang M and colleagues demonstrated that CBS expression was suppressed in the cerebral cortex and hippocampus of mice in TBI. Furthermore, at first it gradually decreased, reaching the minimal values, and then increased. H
2S demonstrated dynamic changes in TBI, in parallel with the expression of the key enzyme of its synthesis [
126].
4.2. Exogenous H2S: Between Neuroprotection and Neurodegeneration
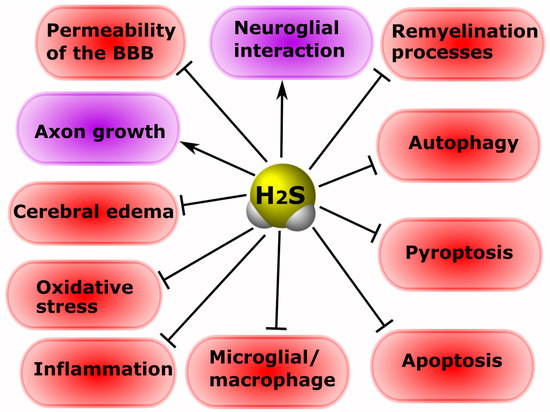
It is known that H
2S is involved in the processes of neuroprotection and neurodegeneration in neurotrauma. The administration of H
2S donors can protect neurons and prevent the development of hemodynamic disorders in TBI [
126]. Increasing the concentration of H
2S reduces cerebral edema, improves motor activity, and reduces apoptosis and autophagy in an animal model of TBI [
133]. According to Jiang and co-authors, H
2S leads to the activation of antioxidant enzymes, reducing the oxidative damage to nervous tissue cells in TBI [
134]. The authors of another study indicate that H
2S reduces mitochondrial dysfunction and autophagy in TBI (
Figure 4) [
135].
Figure 4. The role of H2S in neuroprotection and neurodegeneration in neurotrauma. Arrows with a sharp end—positive regulation; arrows with a blunt end—negative regulation.
Increasing the concentration of H
2S by using a ferrofluid hydrogel (FFH) with iron tetrasulfide (Fe
3S
4) significantly reduces activated microglial/macrophage levels and the expression of pro-inflammatory factors, and increases the rate of directional growth of axons in animal models of SCI [
136]. The use of H
2S-releasing silk fibroin hydrogel resulted in a decrease in the level of neuronal pyroptosis induced by TBI [
137]. NaHS has been reported to reduce the area of spinal cord infarction in the ischemic-reperfusion model of injury [
138].
In a mouse model of sciatic nerve damage, H
2S was shown to significantly reduce neuropathic pain [
145]. In addition, CSE and MST have been found to be present in normal nerves, and axotomy activates CSE in Schwann cells [
113]. Inhibition of H
2S production has been reported to improve the growth of regenerating axons and remyelination processes in peripheral nerve injuries (
Figure 4) [
146].
5. The Role of H2S in Cell Death in Neurotrauma
5.1. Participation of H2S in Oxidative Stress
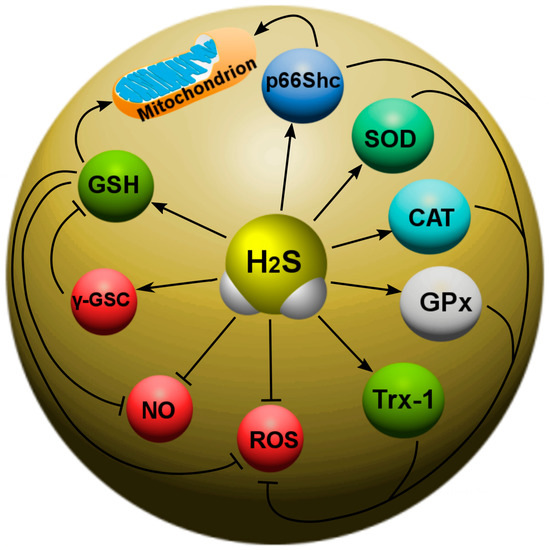
Recently, the main H2S-dependent biological effects in various neurotraumas are considered in the context of the regulation of oxidative stress. It is known that, at 37 °C and a pH of 7.4, more than 80% of H2S molecules dissolve in surface waters and dissociate into the ions H+, HS- and S2−. HS− is a powerful one-electron chemical reagent that effectively traps reactive oxygen species (ROS). Hydrosulfide anions are able to quench ROS by transferring a hydrogen atom or a single electron. The rate of this reaction is directly limited by diffusion. In this case, the reaction of hydrosulfide anions with molecular oxygen proceeds faster in the presence of divalent metal ions. H2S effectively interacts with hypochlorous acid (HClO), hydrogen peroxide (H2O2), lipid hydroperoxides and peroxynitrite (ONOO−), neutralizing their oxidative potential [147]. In addition, H2S itself is a reducing agent that can directly react and extinguish the superoxide anion (O2−), NO and its free radical products, as well as other ROS (Figure 5).
Figure 5. The role of H2S in oxidative stress in neurotrauma. H2S can directly react with and quench ROS and NO. In addition, H2S can increase the level of intracellular reduced glutathione (GSH), which is an antioxidant. However, H2S can activate γ-glutamylcysteine synthase (γ-GSC), which limits GSH synthesis. H2S is involved in the activation of a number of antioxidant defense enzymes: γ-glutamylcysteine synthase (γ-GSC), thioredoxin (Trx-1), superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), and p66Shc.
In addition, H
2S can react with NO to form nitroxide (HNO), which is able to bind to the thiol groups of proteins, leading to the formation of disulfide bonds. HNO can modify GSH with the formation of GSH disulfide and sulfinamide, which can increase oxidative stress and inflammatory processes [
149].
H
2S increases thioredoxin (Trx-1), which is a small (12 kDa) molecule containing the characteristic Cys–Gly–Pro–Cys motif, and the oxidation–reduction of Trx-1 occurs from two of its cysteine residues. Trx-1 is a 12 kDa oxidoreductase enzyme containing a dithiol-disulfide active site that acts as an antioxidant, facilitating the reduction of other proteins by cysteine-thiol-disulfide [
69].
H
2S can bind to the copper (Cu) catalytic center of superoxide dismutase (SOD), which leads to an increase in the rate of absorption of superoxide anions [
151]. Recent studies have also shown that H
2S can attenuate oxidative stress by increasing the activity of catalase (CAT) and glutathione peroxidase (GPx) (
Figure 5).
5.2. Modulation of the H2S Activity of NMDARs and Intracellular Ca2+ Homeostasis
H
2S has been found to modulate NMDAR activity. Protein kinase A (PKA) is known to regulate NMDAR activity. Studies have shown that H
2S can increase levels of cAMP, which, as a secondary messenger, activates PKA. As a result, the activity of the NMDAR increases. However, H
2S can activate NMDARs in an independent way. Since NMDARs are extremely sensitive to oxidation and reduction reactions, the biological effects of H
2S on these receptors may be due to the reduction of disulfide bonds [
118]. H
2S can directly interact with the cysteine residues of receptor subunits, modifying them by S-sulfhydration [
119].
H
2S can increase cytosolic Ca
2+ in neurons by activating slow Ca
2+ L-type channels. Ca
2+ L-type channels are one of the main members of the family of potential-controlled calcium channels. The discovery of these channels occurs in response to a strong depolarization of the membrane and causes a prolonged current of Ca
2+. Ca
2+ L-type channels are expressed in many tissues, including the nervous system [
152]. It is known that these Ca
2+ channels are involved in the pathogenesis of various injuries of the central nervous system and the PNS [
153,
154,
155]. Thus, it has been shown that H
2S can increase the Ca
2+ current in astrocytes, microglia and neurons through the activation of Ca
2+ L-type channels [
156,
157].
5.3. Anti- and Pro-Inflammatory Effects of H2S
Neuro inflammation is an inflammatory response in the nervous tissue characterized by the activation of glial cells, the involvement of neutrophils and macrophages, and the increased synthesis of cytokines, chemokines, free radicals and secondary messengers. The neuroinflammatory response is characterized by a growing front of molecular cellular events underlying secondary damage to nervous tissue [
162,
163]. A number of studies have shown that H
2S plays an important role in inflammatory processes in various pathological conditions, including neurotrauma. The use of ATB-346 (2-(6-methoxynapthalen-2-yl)-propionic acid 4-thiocarbamoyl-phenyl ester), a new H
2S-releasing derivative of naproxen, in TBI, significantly reduced the inflammatory response, due to the inhibition of oxidative stress, nuclear NF-κB (factor kappa-light-chain-enhancer of activated B cells), leukocyte adhesion to the endothelium, tumor necrosis factor (TNF), and interleukin-1 β (IL-1 β) [
67].
The family NF-κB is known to consist of transcription factors that play a complex role in immunity and inflammation. NF-κB regulates inflammation through nuclear translocation followed by the expression of pro-inflammatory factors [
166]. H
2S can modulate the activity of NF-κB activity through trans-sulfonation mechanisms, resulting in the inhibition of the nuclear translocation of NF-κB and a reduced inflammatory response [
167,
168].
5.4. The Effect of H2S on the Level of Neurotrophic Factors
It is known that H2S is able to modulate the level of neurotrophic factors in normal and pathological conditions [26,179,180]. Thus, in a mouse TBI model, the administration of an H2S donor restored GDNF and NGF levels in damaged neural tissue, preserving their neuroprotective effects [67]. The use of a mitochondria-targeted H2S donor in middle cerebral artery occlusion has been reported to increase BDNF and NGF expression, reducing ischemic neuronal damage [26]. The administration of NaHS, a donor of H2S, increased BDNF levels, probably through activation of the transcription factor cAMP response element-binding protein (CREB), which regulates the gene for this neurotrophic factor in brain damage [181].
5.5. Effects of H2S on the Blood-Brain Barrier and Cerebral Edema
Damage to the BBB is the most important pathological substrate of neurotrauma. It was found that H
2S can participate in the restoration of the functional and anatomical integrity of the BBB [
182,
183], as well as reduce cerebral edema [
25,
184]. Thus, in a rat TBI model, it was shown that the use of NaHS, the classic H
2S donor, reduced the excessive permeability of the BBB by activating the mitochondrial adenosine triphosphate-sensitive potassium channels and reducing oxidative stress. Positive H
2S-dependent effects may be associated with the inhibition of PKC-α, β I, β II and δ and the activation of PKC-ε, as well as increased levels of Claudin-5, Occludin and ZO-1 [
185].
5.6. The Role of H2S in Remyelination Processes
Remyelination is an important aspect of the recovery of damaged neurons in injuries of the brain [
187], spinal cord [
188], and peripheral nerves [
189]. The processes associated with demyelination develop as a result of secondary damage, leading to a dysfunction of the neuronal network, neurodegeneration, and ultimately, to the death of neurons [
188]. It is known that H
2S can participate in this process [
146].
It has been shown that the production of H
2S in Schwann cells can lead to destruction of the myelin sheath and the recruitment of macrophages. H
2S positively influences the dedifferentiation and proliferation of Schwann cells in Wallerian degeneration by regulating lysosomal-associated membrane protein 1 (LAMP1), p75 neurotrophin receptor (p75 NTR), c-Jun, and p-ERK1/2. The authors of the study suggest that inhibition of CSE expression may be a potential target in the treatment of pathological processes associated with demyelination [
146].
5.7. H2S-Associated Anti- and Pro-Apoptotic Signaling Mechanisms
H
2S can modulate the apoptosis of neurons and glial cells in neurotrauma. It can act as an anti- and pro-oxidant, as described above, and can regulate the levels of anti- and pro-apoptotic groups of proteins in various traumatic injuries of the nervous system [
29,
30,
31,
133]. H
2S can directly interact with proteins through S-sulfhydration or persulfhydration of cysteine residues on proteins [
190], and bind to metalloproteins, modulating their activity and function [
191]. In addition, H
2S can realize its activity through the activation and inhibition of various signaling pathways [
25,
133].
In a recent study, it was shown that H
2S can inhibit the expression of the p53 protein in damaged neurons, exerting a neuroprotective effect in TBI. Moreover, as the authors suggest, this H
2S-dependent effect was mediated through the pathway p53/glutaminase 2. The use of an H
2S donor showed a significant decrease in TUNEL-positive neuronal and glial cells [
29]. However, in other studies, H
2S caused an increase in p53 expression and the initiation of apoptosis [
39,
40]. Another major pro-apoptotic protein, caspase-3, is also a target for H
2S. Caspase-3 plays a central role in the cascade of caspases, proteolytic enzymes that sequentially activate each other and underlying proteases [
201,
202]. H
2S has been shown to reduce the expression of caspase-3 in damaged neurons and their apoptosis in TBI [
29,
50]. H
2S reduced levels of this proapoptotic enzyme in spinal cord injury models (
Figure 7) [
35,
140].
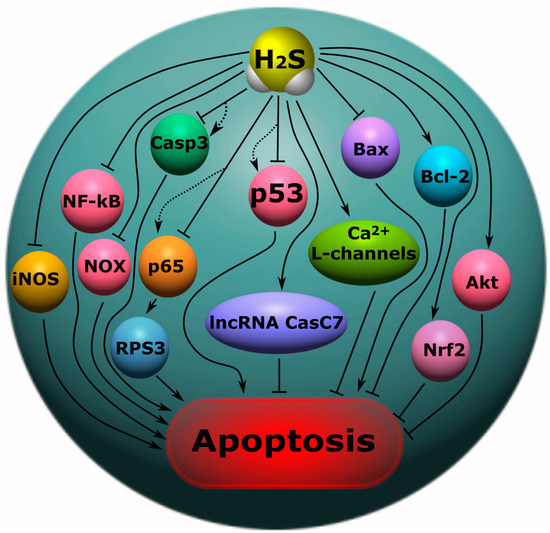
Figure 7. The role of H
2S in apoptosis in neurotrauma. NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; iNOS, inducible nitric oxide synthase; NO, nitric oxide; Casp1, caspase-1; Casp3, caspase-3; NOX, NADPH-oxidase; Bcl-2, B-cell lymphoma 2; Bax, bcl-2-like protein 4; Akt, protein kinase B; p53, tumor protein p53; Nfr2, nuclear factor erythroid 2–related factor 2; p65, RelA; RPS3, ribosomal protein S3; lncRNA CasC7, long non-coding RNA CasC7. Arrows with a sharp end—positive regulation; arrows with a blunt end—negative regulation; dotted line—alternative regulation.
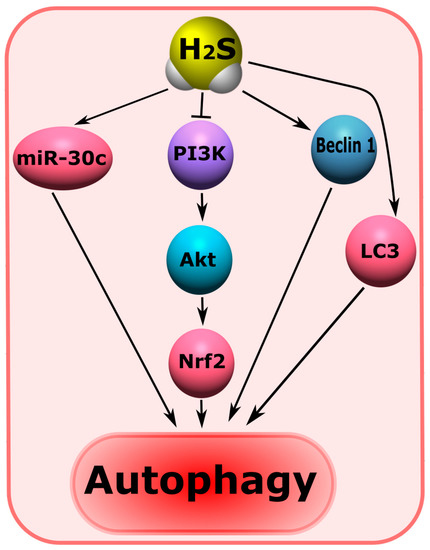
5.8. H2S-Associated Mechanisms of Autophagy
Autophagy plays an important role in the survival and death of neurons in brain injuries [
214]. H
2S has been shown to regulate autophagy-dependent cell death after TBI [
25,
50]. One of the mechanisms for blocking autophagic neuronal death in neurotrauma may be the H
2S-dependent modulation of the PI3K/Akt/Nrf2 pathway and a reduction in oxidative stress [
215]. It is known that PI3K/Akt/Nrf2 is a central mechanism involved in autophagy (
Figure 8) [
216,
217].
Figure 8. The role of H
2S in the regulation of autophagy in neurotrauma. miR-30c, micro-RNA 30c; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B; Nfr2, nuclear factor erythroid 2–related factor 2; Beclin, the mammalian orthologue of yeast Atg6; LC3, Microtubule-associated protein 1A/1B-light chain 3. Arrows with a sharp end—positive regulation; arrows with a blunt end—negative regulation.
5.9. H2S-Associated Mechanisms of Ferroptosis
H
2S plays an important role in ferroptosis. This gasotransmitter can inhibit the process of ferroptosis by increasing the level of antioxidant enzymes, such as GSH, and via ROS uptake [
222]. There are practically no data on the functions of H
2S in the ferroptosis of neurons and glial cells in nervous tissues. For example, H
2S protects the retinal-blood-brain barrier through the activation of the NRF2/KEAP1 signaling pathway, and via AMPK to p62 phosphorylation [
223]. H
2S is reported to protect BV2 microglial cells by reducing lactate dehydrogenase levels (LDH), oxidative stress, lipid peroxidation, and Fe
2+ accumulation [
224].
5.10. H2S-Associated Mechanisms of Pyroptosis
Pyroptosis is a type of programmed necrotic cell death that occurs as a result of caspase-1 activation and the disruption of the integrity of the plasma membrane. A feature of this cell death is the active release of IL-1β and IL-18, dependent on caspase-1, which determines the development of an inflammatory reaction [
225]. To date, it has been proven that pyroptosis plays an important role in the pathogenesis of injuries of the brain and spinal cord [
226]. It has been shown that H
2S may be a key regulator of the processes associated with pyroptosis [
164].
6. The Role of H2S in Mental Disorders and Neurodegenerative Diseases
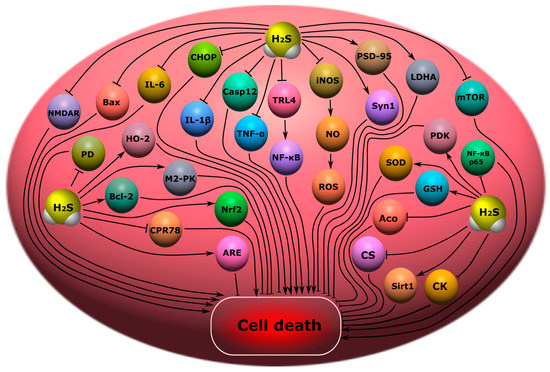
6.1. Cognitive Impairment
Endogenous H
2S may reduce cognitive impairment through the reduction of endoplasmic reticulum stress and the inhibition of caspase-12 and C/EBP homologous protein (CHOP) levels [
27]. It has been shown in a rat model of subarachnoid hemorrhage that H
2S can reduce cognitive deficits by inhibiting the neuroinflammation induced by the TLR4/NF-κB signaling pathway that activates microglial cells (
Figure 9) [
228].
Figure 9. Possible H2S-dependent signaling mechanisms that regulate cell death in the nervous tissue in cognitive impairment and encephalopathy. H2S, hydrogen sulfide; CHOP, C/EBP homologous protein; Casp12, caspase-12; TRL4, toll like receptor 4; NF-ĸB, nuclear factor kappa-light-chain-enhancer of activated B cells; iNOS, inducible nitric oxide synthase; NO, nitric oxide; ROS, reactive oxygen species; PSD-9, postsynaptic density protein 95; Bax, bcl-2-like protein 4; NMDAR, N-methyl-D-aspartate receptor; IL-6, interleukin-6; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α; Syn1, synapsin 1; LDHA, lactate dehydrogenase A; mTOR, mammalian target of rapamycin; PDK, pyruvate dehydrogenase kinase 1; SOD, superoxide dismutase; GSH, glutathione; Aco, aconitase; CS, citrate synthase; Sirt1, NAD-dependent deacetylase sirtuin-1; CK, creatine kinase; NF-ĸB p65, RelA; PD, pyruvate dehydrogenase; Bcl-2, B-cell lymphoma 2; HO-2, heme oxygenase 2; M2-PK, pyruvate kinase M2; CPR78, cuticular protein RR-2 motif 78; Nrf2, nuclear factor erythroid 2–related factor 2; ARE, antioxidant response element.
NaHS, the classic H
2S donor, had a beneficial effect on the memory of TBI-surviving rats [
4]. In a mouse model of surgical trauma accompanied by neuroinflammation, H
2S improved orientation in the Morris water maze. At the same time, the neuroprotective effect of H
2S was due to a decrease in the level of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 in blood serum and in hippocampal cells, which is a key structure of learning and memory [
229].
6.2. Encephalopathy
H
2S can significantly attenuate oxidative stress and apoptosis in encephalopathy through activation of the Nrf2/ARE signaling pathway [
236]. However, a high content of H
2S can lead to a decrease in the activity of citrate synthase (CS) and aconitase (Aco) in the mitochondria of neurons of the cerebral cortex, as well as creatine kinase (CK) in this brain structure, the striatum, and the hippocampus in encephalopathy.
6.3. Depression and Anxiety Disorders
Studies have shown that the administration of NaHS for a week had an antidepressant and anxiolytic effect on mice and rats in various test procedures: forced swimming, tail hanging, and plus maze [
240]. In a model of sleep deprivation in rats, H
2S attenuated depressive and anxiety disorder through the increased expression of Sirt1 in the hippocampus and decreased levels of pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) and CC motif chemokine ligand 2 (CCL2). At the same time, H
2S increased the expression of IL-4 and IL-10, which belong to the anti-inflammatory group of cytokines. The use of the Sirt1 inhibitor leveled the neuroprotective effects of H
2S [
241].
6.4. Epilepsy
In a mouse model of electrically stimulated epileptic seizures, it has been shown that acute and recurrent seizures lead to a decrease in plasma H
2S levels. The authors of the study suggest that H
2S can be considered as a new candidate for the role of a biomarker of severe epileptic seizures. The level of thiocyanate, which is a product of cyanide metabolism via the trans-sulfonation pathway involving H
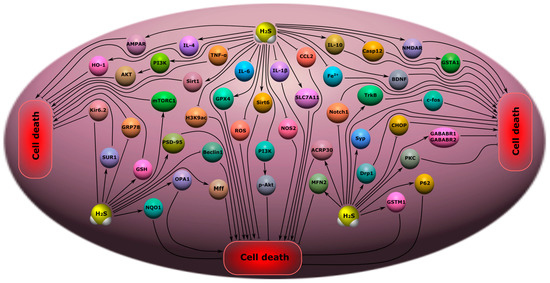
2S, was also strongly reduced in the brain and plasma after convulsions. It is noted that the level of GSH did not change. The study yielded important data on the expression of a number of proteins. An increase in the anti-apoptotic protein optic atrophy 1 (OPA1) was observed, as well as mitochondrial fission factor (Mff), mitofusin 2 (MFN2), and dynamin-1-like protein (Drp1) after epileptic seizures (
Figure 10) [
250].
Figure 10. Possible H2S-dependent signaling mechanisms that regulate cell death in nervous tissue in depression, anxiety disorders, epilepsy, and chronic pain. H2S, hydrogen sulfide; AMPAR, AMPA-type glutamate receptor; IL-4, interleukin-4; IL-6, interleukin-6; IL-1β, interleukin-1β; IL-10, interleukin-10; TNF-α, tumor necrosis factor-α; HO-2, heme oxygenase 2; PI3K, phosphatidylinositol 3-kinase; AKT, RAC-alpha serine/threonine-protein kinase; Sirt1, NAD-dependent deacetylase sirtuin-1; mTORC1, mammalian target of rapamycin complex 1; H3K9ac, acetylated histone H3 lysine 9; GPX4, Glutathione peroxidase 4; Kir6.2, major subunit of the ATP-sensitive K+ channel; SUR1, subunit of the ATP-sensitive K+ channel; GRP78, glucose-regulated protein 78; GSH, glutathione; PSD-9, postsynaptic density protein 95; NQO1, NAD(P)H quinone dehydrogenase 1; OPA1, optic atrophy 1; Beclin, mammalian orthologue of yeast Atg6; Mff, mitochondrial fission factor; ROS, reactive oxygen species; Sirt6, NAD-dependent deacetylase sirtuin-6; p-Akt, phosphorylated RAC-alpha serine/threonine-protein kinase; NOS2, inducible nitric oxide synthase; SLC7A11, solute carrier family 7 member 11; CCL2, C-C motif ligand 2; Fe2+, iron ion; Casp12, caspase-12; NMDAR, N-methyl-D-aspartate receptor; GSTA1, glutathione S-transferase A1; BDNF, brain-derived neurotrophic factor; TrkB, tropomyosin receptor kinase B; c-fos, gene encoding c-fos protein; Notch1, Notch homolog 1; ACRP30, Adipocyte complement-related protein of 30 kDa; MFN2, Mitofusin-2; Syp, synaptophysin; Drp1, dynamin-related protein; CHOP, C\EBP homologous protein; PKC, protein kinase C; P62, sequestosome 1; GSTM1, glutathione s-transferase Mu 1; GABABR1\GABABR2, gamma-aminobutyric acid receptor subunits, GABABR1 and GABABR2.
7. Neurodegenerative Diseases
7.1. Alzheimer’s Disease
Studies have shown that there is a significant decrease in the level of H
2S in the brain of patients suffering from AD [
28]. Violation of H
2S-homeostasis towards a decrease in the level of H
2S is also observed in neurotrauma [
126], which indicates the general mechanisms of dysfunction of the H
2S-synthesizing system in these pathological conditions. H
2S and its metabolites have been proposed as markers of cognitive impairment and vascular dysfunction in AD [
120].
H
2S is reported to inhibit the hyperphosphorylation of Tau by sulfhydration glycogen synthase kinase 3β (GSK3β), improving the motor and cognitive impairment caused by AD [
41]. In a triple transgenic mouse model of AD (3×Tg-AD) demonstrating both Aβ and Tau disorders, the treatment for three months with H
2S significantly protected learning and memory in 3×Tg-AD mice. At the same time, a decrease in amyloid β-plaques in the cortex and hippocampus was observed. These neuroprotective effects were due to the H
2S-dependent downregulation of c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinases, and p38, which play a key role in phosphorylation, Tau, inflammatory response, oxidative stress, and Ca
2+-excitotoxicity (
Figure 11) [
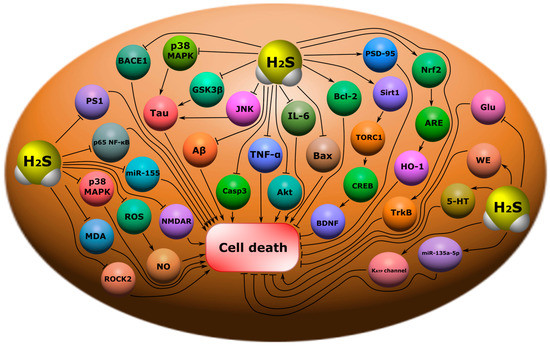
263].
Figure 11. Possible H
2S-dependent signaling mechanisms that regulate cell death in nervous tissue in neurodegenerative diseases. BACE1, beta-site amyloid precursor protein cleaving enzyme 1; p38 MAPK, p38 mitogen-activated protein kinase; Tau, microtubule-associated protein tau; GSK3β, glycogen synthase kinase-3 beta; JNK, c-Jun N-terminal kinase; Aβ, amyloid beta; Casp3, caspase-3; TNF-α, tumor necrosis factor-α; Akt, RAC-alpha serine/threonine-protein kinase; IL-6, interleukin-6; Bax, bcl-2-like protein 4; Bcl-2, B-cell lymphoma 2; PSD-95, postsynaptic density protein 95; Sirt1, NAD-dependent deacetylase sirtuin-1; TORC1, target of rapamycin kinase complex 1; CREB, cAMP response element-binding protein; BDNF, brain-derived neurotrophic factor; Nrf2, nuclear factor erythroid 2–related factor 2; ARE, antioxidant response element; HO-1, heme oxygenase 1; TrkB, tropomyosin receptor kinase B; Glu, glutamic acid; WE, Warburg effect; 5-HT, serotonin; miR-133a-5p, microRNA 133a-5p; K
ATP channel, ATP-sensitive K
+ channel; PS1, presenilin-1; p65 NF-ĸB, nuclear factor kappa-light-chain-enhancer of activated B cells; miR-155, microRNA 155; ROS, reactive oxygen species; NO, nitric oxide; MDA, malonic dialdehyde; NMDAR, N-methyl-D-aspartate receptor; ROCK2, Rho associated coiled-coil containing protein kinase 2.
7.2. Parkinson’s Disease
Abnormal levels of H
2S and its metabolites have been identified in many studies in PD. Thus, it was shown that the level of endogenous H
2S significantly decreased in the substantia nigra (SN) in PD, and the use of H
2S donors contributed to the neuroprotective effect and reduced the death of dopaminergic neurons in this pathological condition [
270]. The protective effect of H
2S has been shown in various PD models in vitro and in vivo [
271].
It was shown that the use of the H
2S donor ACS84, a derivative of the compound L-Dopa, reduced motor dysfunction in mice with induced PD by reducing neuronal loss in the SN, via Nrf-2 activation, and through the increased expression of a number of antioxidant enzymes [
272].
8. Therapeutic Approaches Using H2S as a Neuroprotector
Many experimental animal studies have shown that H2S has a beneficial effect on the survival of neurons and glial cells in various pathological conditions, including neurotrauma, and psychiatric and neurodegenerative diseases. However, there is no H2S-associated neuroprotector that has undergone clinical trials, yet.
The complexity of the development of this drug lies in the fact that it is necessary to understand the molecular signaling mechanisms associated with H
2S well, which are realized under the conditions of a particular pathological condition. This is not the end of the problem. The therapeutic effect of H
2S largely depends on its concentration: a level of H
2S close to the physiological level exhibits a neuroprotective effect, and a high level of H
2S can lead to cytotoxicity. Therefore, this drug should release H
2S at the required concentration for a long time and have a neurocumulative effect in order to accumulate precisely in the nervous tissue and not release H
2S in non-target organs. Moreover, if there are already good developments on the first point, then the situation is more complicated regarding the second [
279].
However, the situation with slow H
2S donors is also ambiguous. For example, GYY4137 is reported to be ineffective due to the low release of H
2S, requiring high concentrations. In this connection, new, long-acting H
2S donors based on GYY4137, such as AP67 and AP105, have been developed. They exhibit high biological activity [
284]. FW1256 could be a promising H
2S donor; it has demonstrated a good anti-inflammatory effect in a number of studies [
285].
9. Conclusions
Injuries of the central and peripheral nervous system and associated neurodegenerative diseases and mental disorders are one of the main causes of disability and death worldwide after cardiovascular and oncological diseases. H2S can be considered as a potential molecular target for neuroprotective effects. Definitely, the positive role of H2S, manifested in the reduction of oxidative stress, inflammation, demyelination processes, excitotoxicity, apoptosis, autophagy, ferroptosis, and pyroptosis, prevail over its negative effects in the nervous tissue during traumatic damage to the CNS and PNS. In addition, the use of H2S donors effectively reduces the symptoms of mental disorders, such as cognitive impairment, encephalopathy, depression and anxiety disorders, epilepsy, chronic pain, and also inhibits the progression of neurodegenerative diseases.