 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Farhad Bazgir | -- | 4234 | 2023-07-12 14:28:00 | | | |
| 2 | Lindsay Dong | Meta information modification | 4234 | 2023-07-14 02:44:25 | | |
Video Upload Options
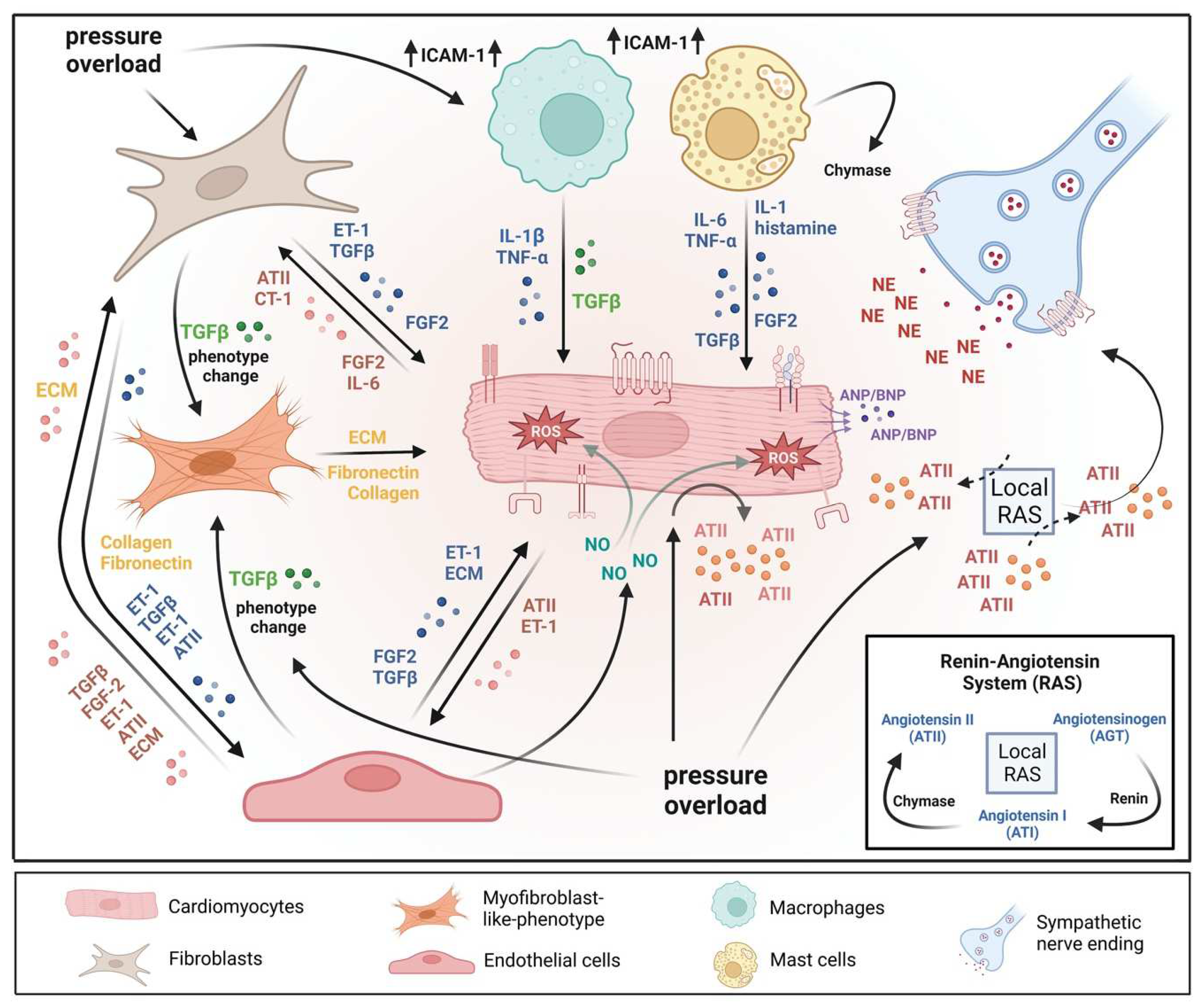
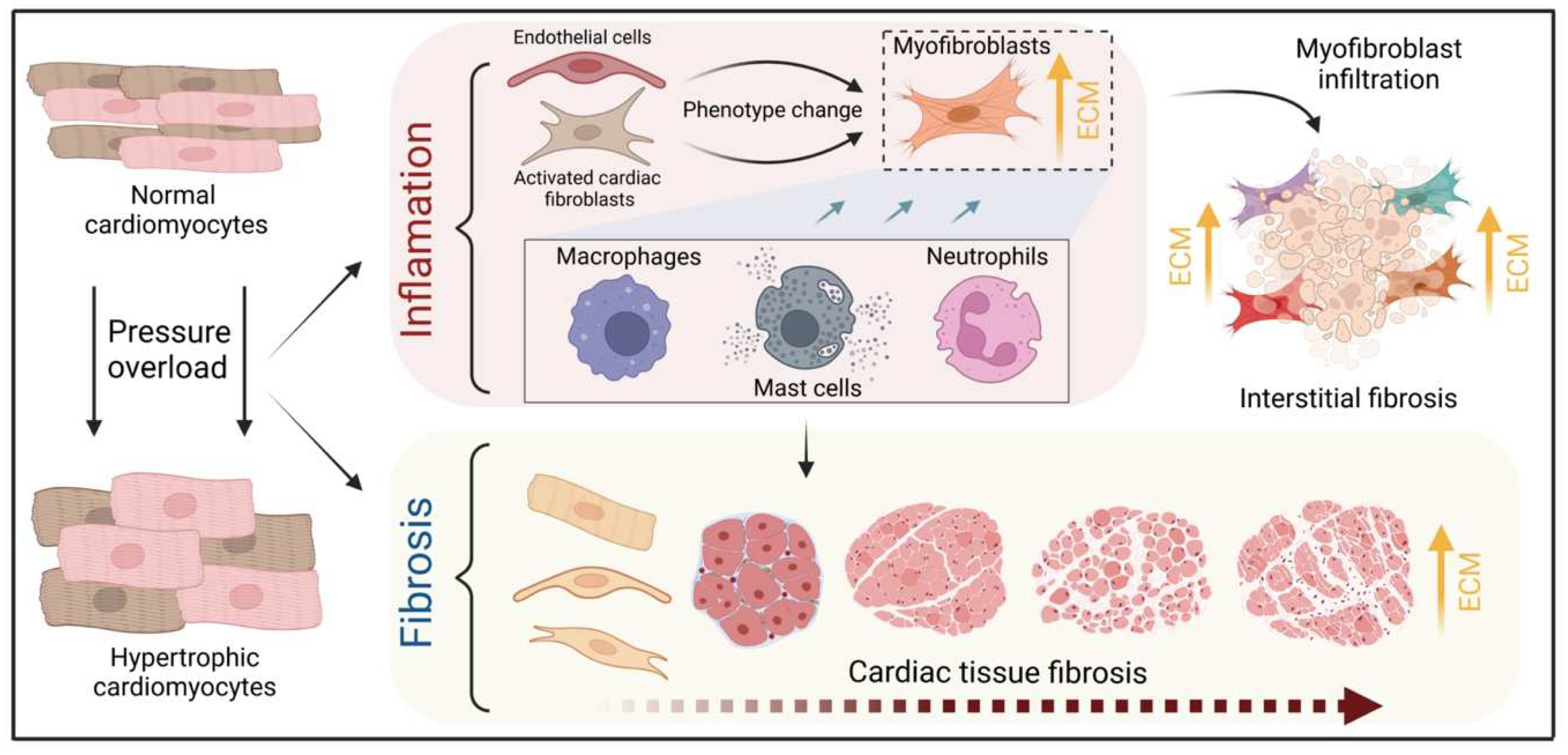
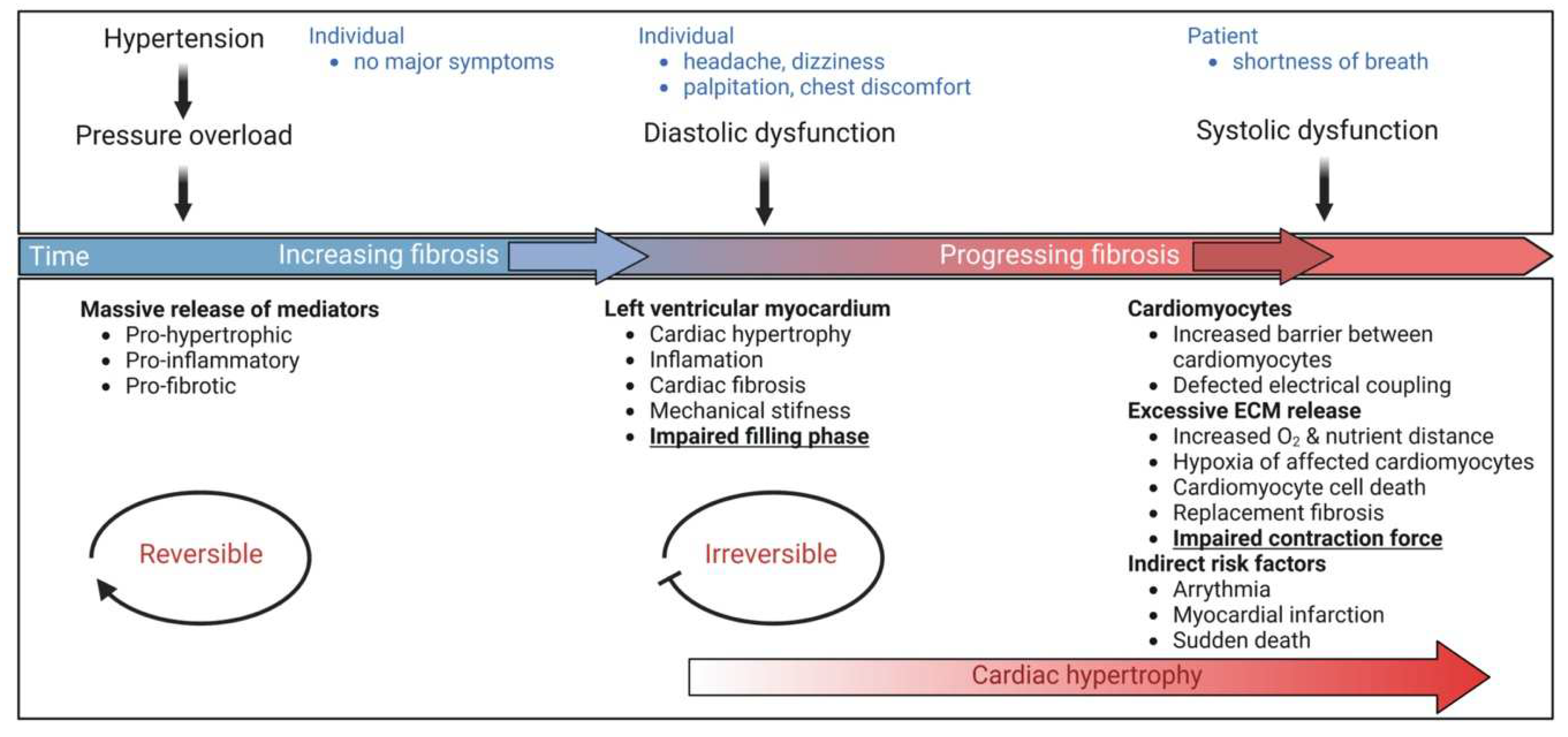
Pathological cardiac hypertrophy is a key risk factor for the development of heart failure and predisposes individuals to cardiac arrhythmia and sudden death. While physiological cardiac hypertrophy is adaptive, hypertrophy resulting from conditions comprising hypertension, aortic stenosis, or genetic mutations, such as hypertrophic cardiomyopathy, is maladaptive. Prolonged cardiovascular stress causes cardiomyocytes and non-myocardial cells to enter an activated state releasing numerous pro-hypertrophic, pro-fibrotic, and pro-inflammatory mediators such as vasoactive hormones, growth factors, and cytokines, i.e., commencing signaling events that collectively cause cardiac hypertrophy. Fibrotic remodeling is mediated by cardiac fibroblasts as the central players, but also endothelial cells and resident and infiltrating immune cells enhance these processes. Many of these hypertrophic mediators are now being integrated into computational models that provide system-level insights and will help to translate our knowledge into new pharmacological targets.
1. General Introduction
2. An Interplay of Different Cells in Hypertrophic Remodeling
2.1. Fibroblast Remodeling
2.2. Endothelial Cell Activation
3. The Role of Immune Cells in Cardiac Hypertrophy
3.1. Cardiac Mast Cells
3.2. Monocytes & Macrophages
3.3. Neutrophils
3.4. Lymphocytes
3.5. Sympathetic Neurons
4. Mediators of Cardiac Remodeling
4.1. Activation of the Local Renin-Angiotensin System (RAS)
4.2. Reactive Oxygen Species (ROS)
4.3. Endogenous Storage Pools of AT-II in Secretory Granules
4.4. The Two Faces of the TGF-ß Signaling
4.5. Endothelin-1 Effects
4.6. FGF-2 Effects in Scar Formation
4.7. Cytokines and Inflammasome in Cardiac Remodeling
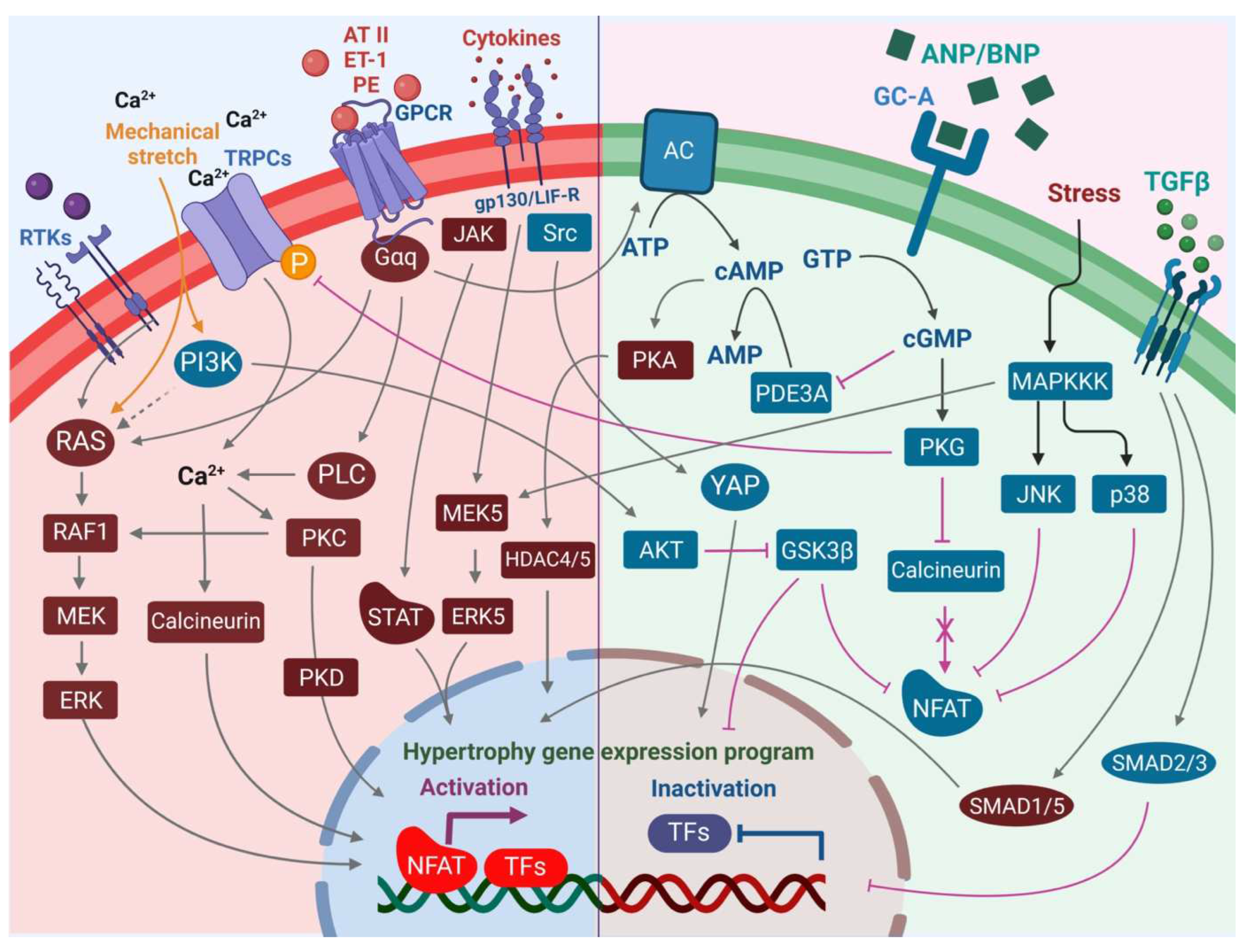
4.8. Calcineurin/NFAT in Cardiac Hypertrophy
4.9. ANP/BNP in Cardiac Hypertrophy
5. Mathematical Modeling of Cardiac Remodeling
5.1. Computational Models of Cardiac Hypertrophy
5.2. Computational Modeling of Fibrosis
References
- Zhu, L.; Li, C.; Liu, Q.; Xu, W.; Zhou, X. Molecular biomarkers in cardiac hypertrophy. J. Cell. Mol. Med. 2019, 23, 1671–1677.
- Schaub, M.C.; Hefti, M.A.; Harder, B.A.; Eppenberger, H.M. Various hypertrophic stimuli induce distinct phenotypes in cardiomyocytes. J. Mol. Med. 1997, 75, 901–920.
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600.
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48.
- Ovchinnikova, E.; Hoes, M.; Ustyantsev, K.; Bomer, N.; de Jong, T.V.; van der Mei, H.; Berezikov, E.; van der Meer, P. Modeling Human Cardiac Hypertrophy in Stem Cell-Derived Cardiomyocytes. Stem Cell Rep. 2018, 10, 794–807.
- Berenji, K.; Drazner, M.H.; Rothermel, B.A.; Hill, J.A. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H8–H16.
- Haider, A.W.; Larson, M.G.; Benjamin, E.J.; Levy, D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J. Am. Coll. Cardiol. 1998, 32, 1454–1459.
- Nakhaei-Rad, S.; Bazgir, F.; Dahlmann, J.; Busley, A.V.; Buchholzer, M.; Haghighi, F.; Schänzer, A.; Hahn, A.; Kötter, S.; Schanze, D. Alteration of myocardial structure and function in RAF1-associated Noonan syndrome: Insights from cardiac disease modeling based on patient-derived iPSCs. bioRxiv 2022.
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314.
- Zhang, M.; Shah, A.M. ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 2014, 102, 249–257.
- Ding, S.; Wang, D.; Zhou, X.; Chen, L.; Feng, K.; Xu, X.; Huang, T.; Li, Z.; Cai, Y. Predicting heart cell types by using transcriptome profiles and a machine learning method. Life 2022, 12, 228.
- Peter, A.K.; Bjerke, M.A.; Leinwand, L.A. Biology of the cardiac myocyte in heart disease. Mol. Biol. Cell 2016, 27, 2149–2160.
- Takeda, N.; Manabe, I. Cellular Interplay between Cardiomyocytes and Nonmyocytes in Cardiac Remodeling. Int. J. Inflamm. 2011, 2011, 535241.
- Hefti, M.A.; Harder, B.A.; Eppenberger, H.M.; Schaub, M.C. Signaling pathways in cardiac myocyte hypertrophy. J. Mol. Cell. Cardiol. 1997, 29, 2873–2892.
- Nikolov, A.; Popovski, N. Extracellular matrix in heart disease: Focus on circulating collagen type I and III derived peptides as biomarkers of myocardial fibrosis and their potential in the prognosis of heart failure: A concise review. Metabolites 2022, 12, 297.
- Moore-Morris, T.; Guimaraes-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934.
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; J Lin, S.-C.; Aronow, B.J.; Tallquist, M.D. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260.
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491.
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363.
- Sun, C.; Tian, X.; Jia, Y.; Yang, M.; Li, Y.; Fernig, D.G. Functions of exogenous FGF signals in regulation of fibroblast to myofibroblast differentiation and extracellular matrix protein expression. Open Biol. 2022, 12, 210356.
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961.
- Cheng, W.; Li, X.; Liu, D.; Cui, C.; Wang, X. Endothelial-to-mesenchymal transition: Role in cardiac fibrosis. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 3–11.
- Xu, X.; Tan, X.; Tampe, B.; Nyamsuren, G.; Liu, X.; Maier, L.S.; Sossalla, S.; Kalluri, R.; Zeisberg, M.; Hasenfuss, G.; et al. Epigenetic balance of aberrant Rasal1 promoter methylation and hydroxymethylation regulates cardiac fibrosis. Cardiovasc. Res. 2015, 105, 279–291.
- Brutsaert, D.L. Cardiac endothelial-myocardial signaling: Its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 2003, 83, 59–115.
- Esper, R.J.; Nordaby, R.A.; Vilarino, J.O.; Paragano, A.; Cacharron, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4.
- Kuhn, M. Cardiology: A big-hearted molecule. Nature 2015, 519, 416–417.
- Wang, M.; Li, Y.; Li, S.; Lv, J. Endothelial dysfunction and diabetic cardiomyopathy. Front. Endocrinol. 2022, 13, 851941.
- Gray, M.O.; Long, C.S.; Kalinyak, J.E.; Li, H.T.; Karliner, J.S. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc. Res. 1998, 40, 352–363.
- Drawnel, F.M.; Archer, C.R.; Roderick, H.L. The role of the paracrine/autocrine mediator endothelin-1 in regulation of cardiac contractility and growth. Br. J. Pharm. 2013, 168, 296–317.
- Froogh, G.; Kandhi, S.; Duvvi, R.; Le, Y.; Weng, Z.; Alruwaili, N.; Ashe, J.O.; Sun, D.; Huang, A. The contribution of chymase-dependent formation of ANG II to cardiac dysfunction in metabolic syndrome of young rats: Roles of fructose and EETs. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H985–H993.
- Dvorak, A.M. Mast-cell degranulation in human hearts. N. Engl. J. Med. 1986, 315, 969–970.
- Ingason, A.B.; Mechmet, F.; Atacho, D.A.M.; Steingrímsson, E.; Petersen, P.H. Distribution of mast cells within the mouse heart and its dependency on Mitf. Mol. Immunol. 2019, 105, 9–15.
- Mekori, Y.A.; Metcalfe, D.D. Mast cells in innate immunity. Immunol. Rev. 2000, 173, 131–140.
- Liu, X.; Shi, G.P.; Guo, J. Innate Immune Cells in Pressure Overload-Induced Cardiac Hypertrophy and Remodeling. Front. Cell Dev. Biol. 2021, 9, 659666.
- Balakumar, P.; Singh, A.P.; Ganti, S.S.; Krishan, P.; Ramasamy, S.; Singh, M. Resident cardiac mast cells: Are they the major culprit in the pathogenesis of cardiac hypertrophy? Basic Clin. Pharmacol. Toxicol. 2008, 102, 5–9.
- Shiota, N.; Rysa, J.; Kovanen, P.T.; Ruskoaho, H.; Kokkonen, J.O.; Lindstedt, K.A. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J. Hypertens. 2003, 21, 1935–1944.
- Petrov, V.V.; Fagard, R.H.; Lijnen, P.J. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension 2002, 39, 258–263.
- Weber, K.T. Fibrosis and hypertensive heart disease. Curr. Opin. Cardiol. 2000, 15, 264–272.
- Leurs, R.; Bakker, R.A.; Timmerman, H.; de Esch, I.J. The histamine H3 receptor: From gene cloning to H3 receptor drugs. Nat. Rev. Drug. Discov. 2005, 4, 107–120.
- Hough, L.B. Genomics meets histamine receptors: New subtypes, new receptors. Mol. Pharm. 2001, 59, 415–419.
- Levick, S.P. Histamine receptors in heart failure. Heart Fail. Rev. 2022, 27, 1355–1372.
- Galli, S.J. New concepts about the mast cell. N. Engl. J. Med. 1993, 328, 257–265.
- Bradding, P.; Feather, I.H.; Howarth, P.H.; Mueller, R.; Roberts, J.A.; Britten, K.; Bews, J.P.; Hunt, T.C.; Okayama, Y.; Heusser, C.H.; et al. Interleukin 4 is localized to and released by human mast cells. J. Exp. Med. 1992, 176, 1381–1386.
- Ohkawara, Y.; Yamauchi, K.; Tanno, Y.; Tamura, G.; Ohtani, H.; Nagura, H.; Ohkuda, K.; Takishima, T. Human lung mast cells and pulmonary macrophages produce tumor necrosis factor-alpha in sensitized lung tissue after IgE receptor triggering. Am. J. Respir. Cell. Mol. Biol. 1992, 7, 385–392.
- Azzawi, M.; Kan, S.W.; Hillier, V.; Yonan, N.; Hutchinson, I.V.; Hasleton, P.S. The distribution of cardiac macrophages in myocardial ischaemia and cardiomyopathy. Histopathology 2005, 46, 314–319.
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104.
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295.
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804.
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158.
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969.
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126.
- Hinglais, N.; Heudes, D.; Nicoletti, A.; Mandet, C.; Laurent, M.; Bariety, J.; Michel, J.B. Colocalization of myocardial fibrosis and inflammatory cells in rats. Lab. Investig. 1994, 70, 286–294.
- Nicoletti, A.; Heudes, D.; Mandet, C.; Hinglais, N.; Bariety, J.; Michel, J.B. Inflammatory cells and myocardial fibrosis: Spatial and temporal distribution in renovascular hypertensive rats. Cardiovasc. Res. 1996, 32, 1096–1107.
- Bratton, D.L.; Henson, P.M. Neutrophil clearance: When the party is over, clean-up begins. Trends Immunol. 2011, 32, 350–357.
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.A.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094.
- Bennouna, S.; Bliss, S.K.; Curiel, T.J.; Denkers, E.Y. Cross-talk in the innate immune system: Neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J. Immunol. 2003, 171, 6052–6058.
- Chertov, O.; Ueda, H.; Xu, L.L.; Tani, K.; Murphy, W.J.; Wang, J.M.; Howard, O.M.; Sayers, T.J.; Oppenheim, J.J. Identification of human neutrophil-derived cathepsin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. J. Exp. Med. 1997, 186, 739–747.
- Tsuda, Y.; Takahashi, H.; Kobayashi, M.; Hanafusa, T.; Herndon, D.N.; Suzuki, F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 2004, 21, 215–226.
- Bui, T.A.; Jickling, G.C.; Winship, I.R. Neutrophil dynamics and inflammaging in acute ischemic stroke: A transcriptomic review. Front. Aging Neurosci. 2022, 14, 1041333.
- van Gisbergen, K.P.; Sanchez-Hernandez, M.; Geijtenbeek, T.B.; van Kooyk, Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J. Exp. Med. 2005, 201, 1281–1292.
- Li, J.M.; Gall, N.P.; Grieve, D.J.; Chen, M.; Shah, A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002, 40, 477–484.
- Ciz, M.; Denev, P.; Kratchanova, M.; Vasicek, O.; Ambrozova, G.; Lojek, A. Flavonoids inhibit the respiratory burst of neutrophils in mammals. Oxid. Med. Cell. Longev. 2012, 2012, 181295.
- Ozyilmaz, S.; Akgul, O.; Uyarel, H.; Pusuroglu, H.; Gul, M.; Satilmisoglu, M.H.; Bolat, I.; Ozyilmaz, I.; Uçar, H.; Yildirim, A. The importance of the neutrophil-to-lymphocyte ratio in patients with hypertrophic cardiomyopathy. Rev. Port. Cardiol. 2017, 36, 239–246.
- Fries, R.C.; Kadotani, S.; Stack, J.P.; Kruckman, L.; Wallace, G. Prognostic Value of Neutrophil-to-Lymphocyte Ratio in Cats With Hypertrophic Cardiomyopathy. Front. Vet. Sci. 2022, 9, 813524.
- Cordero-Reyes, A.M.; Youker, K.A.; Trevino, A.R.; Celis, R.; Hamilton, D.J.; Flores-Arredondo, J.H.; Orrego, C.M.; Bhimaraj, A.; Estep, J.D.; Torre-Amione, G. Full expression of cardiomyopathy is partly dependent on B-cells: A pathway that involves cytokine activation, immunoglobulin deposition, and activation of apoptosis. J. Am. Heart Assoc. 2016, 5, e002484.
- Sobirin, M.A.; Kinugawa, S.; Takahashi, M.; Fukushima, A.; Homma, T.; Ono, T.; Hirabayashi, K.; Suga, T.; Azalia, P.; Takada, S. Activation of natural killer T cells ameliorates postinfarct cardiac remodeling and failure in mice. Circ. Res. 2012, 111, 1037–1047.
- Wang, H.-X.; Li, W.-J.; Hou, C.-L.; Lai, S.; Zhang, Y.-L.; Tian, C.; Yang, H.; Du, J.; Li, H.-H. CD1d-dependent natural killer T cells attenuate angiotensin II-induced cardiac remodelling via IL-10 signalling in mice. Cardiovasc. Res. 2019, 115, 83–93.
- Isaka, M.; Kudo, A.; Imamura, M.; Kawakami, H.; Yasuda, K. Endothelin receptors, localized in sympathetic nerve terminals of the heart, modulate norepinephrine release and reperfusion arrhythmias. Basic Res. Cardiol. 2007, 102, 154–162.
- Lehmann, L.H.; Stanmore, D.A.; Backs, J. The role of endothelin-1 in the sympathetic nervous system in the heart. Life Sci. 2014, 118, 165–172.
- Lehmann, L.H.; Rostosky, J.S.; Buss, S.J.; Kreusser, M.M.; Krebs, J.; Mier, W.; Enseleit, F.; Spiger, K.; Hardt, S.E.; Wieland, T.; et al. Essential role of sympathetic endothelin A receptors for adverse cardiac remodeling. Proc. Natl. Acad. Sci. USA 2014, 111, 13499–13504.
- Mackins, C.J.; Kano, S.; Seyedi, N.; Schafer, U.; Reid, A.C.; Machida, T.; Silver, R.B.; Levi, R. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J. Clin. Investig. 2006, 116, 1063–1070.
- Reid, A.C.; Mackins, C.J.; Seyedi, N.; Levi, R.; Silver, R.B. Coupling of angiotensin II AT1 receptors to neuronal NHE activity and carrier-mediated norepinephrine release in myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1448–H1454.
- Rodriguez-Gonzalez, M.; Lubian-Gutierrez, M.; Cascales-Poyatos, H.M.; Perez-Reviriego, A.A.; Castellano-Martinez, A. Role of the Renin–Angiotensin–Aldosterone System in Dystrophin-Deficient Cardiomyopathy. Int. J. Mol. Sci. 2020, 22, 356.
- Campbell, D.J. Circulating and tissue angiotensin systems. J. Clin. Investig. 1987, 79, 1–6.
- Dinh, D.T.; Frauman, A.G.; Johnston, C.I.; Fabiani, M.E. Angiotensin receptors: Distribution, signalling and function. Clin. Sci. 2001, 100, 481–492.
- Lindpaintner, K.; Ganten, D. The cardiac renin-angiotensin system. An appraisal of present experimental and clinical evidence. Circ. Res. 1991, 68, 905–921.
- Baker, K.M.; Booz, G.W.; Dostal, D.E. Cardiac actions of angiotensin II: Role of an intracardiac renin-angiotensin system. Annu. Rev. Physiol. 1992, 54, 227–241.
- Lee, M.A.; Bohm, M.; Paul, M.; Ganten, D. Tissue renin-angiotensin systems. Their role in cardiovascular disease. Circulation 1993, 87, IV7–IV13.
- Sadoshima, J.; Xu, Y.; Slayter, H.S.; Izumo, S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 1993, 75, 977–984.
- Malhotra, R.; Sadoshima, J.; Brosius III, F.C.; Izumo, S. Mechanical stretch and angiotensin II differentially upregulate the renin-angiotensin system in cardiac myocytes in vitro. Circ. Res. 1999, 85, 137–146.
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424.
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res. 2009, 81, 449–456.
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289.
- Carnicer, R.; Crabtree, M.J.; Sivakumaran, V.; Casadei, B.; Kass, D.A. Nitric oxide synthases in heart failure. Antioxid. Redox Signal. 2013, 18, 1078–1099.
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041.
- Zhang, Y.; Tocchetti, C.G.; Krieg, T.; Moens, A.L. Oxidative and nitrosative stress in the maintenance of myocardial function. Free. Radic. Biol. Med. 2012, 53, 1531–1540.
- Nediani, C.; Raimondi, L.; Borchi, E.; Cerbai, E. Nitric oxide/reactive oxygen species generation and nitroso/redox imbalance in heart failure: From molecular mechanisms to therapeutic implications. Antioxid. Redox Signal. 2011, 14, 289–331.
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407.
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673.
- Sadoshima, J.; Izumo, S. The heterotrimeric G q protein-coupled angiotensin II receptor activates p21 ras via the tyrosine kinase-Shc-Grb2-Sos pathway in cardiac myocytes. EMBO J. 1996, 15, 775–787.
- Kala, P.; Gawrys, O.; Miklovič, M.; Vaňourková, Z.; Škaroupková, P.; Jíchová, Š.; Sadowski, J.; Kompanowska-Jezierska, E.; Walkowska, A.; Veselka, J. Endothelin type A receptor blockade attenuates aorto-caval fistula-induced heart failure in rats with angiotensin II-dependent hypertension. J. Hypertens. 2023, 41, 99–114.
- Harada, M.; Itoh, H.; Nakagawa, O.; Ogawa, Y.; Miyamoto, Y.; Kuwahara, K.; Ogawa, E.; Igaki, T.; Yamashita, J.; Masuda, I.; et al. Significance of ventricular myocytes and nonmyocytes interaction during cardiocyte hypertrophy: Evidence for endothelin-1 as a paracrine hypertrophic factor from cardiac nonmyocytes. Circulation 1997, 96, 3737–3744.
- Pellieux, C.; Foletti, A.; Peduto, G.; Aubert, J.F.; Nussberger, J.; Beermann, F.; Brunner, H.R.; Pedrazzini, T. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J. Clin. Investig. 2001, 108, 1843–1851.
- Jiang, Z.S.; Jeyaraman, M.; Wen, G.B.; Fandrich, R.R.; Dixon, I.M.; Cattini, P.A.; Kardami, E. High- but not low-molecular weight FGF-2 causes cardiac hypertrophy in vivo; possible involvement of cardiotrophin-1. J. Mol. Cell. Cardiol. 2007, 42, 222–233.
- Sadoshima, J.; Izumo, S. Molecular characterization of angiotensin II—Induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 1993, 73, 413–423.
- Harada, K.; Komuro, I.; Zou, Y.; Kudoh, S.; Kijima, K.; Matsubara, H.; Sugaya, T.; Murakami, K.; Yazaki, Y. Acute pressure overload could induce hypertrophic responses in the heart of angiotensin II type 1a knockout mice. Circ. Res. 1998, 82, 779–785.
- Harada, K.; Komuro, I.; Shiojima, I.; Hayashi, D.; Kudoh, S.; Mizuno, T.; Kijima, K.; Matsubara, H.; Sugaya, T.; Murakami, K.; et al. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 1998, 97, 1952–1959.
- Border, W.A.; Noble, N.A. Transforming growth factor beta in tissue fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292.
- Yang, Y.C.; Piek, E.; Zavadil, J.; Liang, D.; Xie, D.; Heyer, J.; Pavlidis, P.; Kucherlapati, R.; Roberts, A.B.; Bottinger, E.P. Hierarchical model of gene regulation by transforming growth factor beta. Proc. Natl. Acad. Sci. USA 2003, 100, 10269–10274.
- Verrecchia, F.; Chu, M.L.; Mauviel, A. Identification of novel TGF-beta/Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 2001, 276, 17058–17062.
- Bujak, M.; Ren, G.; Kweon, H.J.; Dobaczewski, M.; Reddy, A.; Taffet, G.; Wang, X.F.; Frangogiannis, N.G. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 2007, 116, 2127–2138.
- Ryer, E.J.; Hom, R.P.; Sakakibara, K.; Nakayama, K.I.; Nakayama, K.; Faries, P.L.; Liu, B.; Kent, K.C. PKCdelta is necessary for Smad3 expression and transforming growth factor beta-induced fibronectin synthesis in vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 2006, 26, 780–786.
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 2002, 106, 130–135.
- Divakaran, V.; Adrogue, J.; Ishiyama, M.; Entman, M.L.; Haudek, S.; Sivasubramanian, N.; Mann, D.L. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circ. Heart Fail. 2009, 2, 633–642.
- Chowdhury, M.A.; Moukarbel, G.V.; Gupta, R.; Frank, S.M.; Anderson, A.M.; Liu, L.C.; Khouri, S.J. Endothelin 1 is associated with heart failure hospitalization and long-term mortality in patients with heart failure with preserved ejection fraction and pulmonary hypertension. Cardiology 2019, 143, 124–133.
- Jankowich, M.; Choudhary, G. Endothelin-1 levels and cardiovascular events. Trends Cardiovasc. Med. 2020, 30, 1–8.
- Kedzierski, R.M.; Yanagisawa, M. Endothelin system: The double-edged sword in health and disease. Annu. Rev. Pharm. Toxicol. 2001, 41, 851–876.
- Hilal-Dandan, R.; Merck, D.T.; Lujan, J.P.; Brunton, L.L. Coupling of the type A endothelin receptor to multiple responses in adult rat cardiac myocytes. Mol. Pharm. 1994, 45, 1183–1190.
- Sano, F.K.; Akasaka, H.; Shihoya, W.; Nureki, O. Cryo-EM structure of the endothelin-1-ETB-Gi complex. Elife 2023, 12, e85821.
- Du, J.; Zheng, L.; Gao, P.; Yang, H.; Yang, W.-J.; Guo, F.; Liang, R.; Feng, M.; Wang, Z.; Zhang, Z. A small-molecule cocktail promotes mammalian cardiomyocyte proliferation and heart regeneration. Cell Stem Cell 2022, 29, 545–558.e13.
- Auchampach, J.; Han, L.; Huang, G.N.; Kühn, B.; Lough, J.W.; O’Meara, C.C.; Payumo, A.Y.; Rosenthal, N.A.; Sucov, H.M.; Yutzey, K.E. Measuring cardiomyocyte cell-cycle activity and proliferation in the age of heart regeneration. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H579–H596.
- Venugopal, H.; Hanna, A.; Humeres, C.; Frangogiannis, N.G. Properties and functions of fibroblasts and myofibroblasts in myocardial infarction. Cells 2022, 11, 1386.
- Rao, Z.; Shen, D.; Chen, J.; Jin, L.; Wu, X.; Chen, M.; Li, L.; Chu, M.; Lin, J. Basic fibroblast growth factor attenuates injury in myocardial infarction by enhancing hypoxia-inducible factor-1 alpha accumulation. Front. Pharmacol. 2020, 11, 1193.
- Detillieux, K.A.; Sheikh, F.; Kardami, E.; Cattini, P.A. Biological activities of fibroblast growth factor-2 in the adult myocardium. Cardiovasc. Res. 2003, 57, 8–19.
- Jimenez, S.K.; Sheikh, F.; Jin, Y.; Detillieux, K.A.; Dhaliwal, J.; Kardami, E.; Cattini, P.A. Transcriptional regulation of FGF-2 gene expression in cardiac myocytes. Cardiovasc. Res. 2004, 62, 548–557.
- Schultz, J.E.; Witt, S.A.; Nieman, M.L.; Reiser, P.J.; Engle, S.J.; Zhou, M.; Pawlowski, S.A.; Lorenz, J.N.; Kimball, T.R.; Doetschman, T. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J. Clin. Investig. 1999, 104, 709–719.
- Morrison, R.S.; Yamaguchi, F.; Saya, H.; Bruner, J.M.; Yahanda, A.M.; Donehower, L.A.; Berger, M. Basic fibroblast growth factor and fibroblast growth factor receptor I are implicated in the growth of human astrocytomas. J. Neurooncol. 1994, 18, 207–216.
- Reed, M.J.; Purohit, A.; Duncan, L.J.; Singh, A.; Roberts, C.J.; Williams, G.J.; Potter, B.V. The role of cytokines and sulphatase inhibitors in regulating oestrogen synthesis in breast tumours. J. Steroid Biochem. Mol. Biol. 1995, 53, 413–420.
- Halaban, R. Growth factors and melanomas. Semin. Oncol. 1996, 23, 673–681.
- Kumar-Singh, S.; Weyler, J.; Martin, M.J.; Vermeulen, P.B.; Van Marck, E. Angiogenic cytokines in mesothelioma: A study of VEGF, FGF-1 and -2, and TGF beta expression. J. Pathol. 1999, 189, 72–78.
- Dow, J.K.; deVere White, R.W. Fibroblast growth factor 2: Its structure and property, paracrine function, tumor angiogenesis, and prostate-related mitogenic and oncogenic functions. Urology 2000, 55, 800–806.
- Auguste, P.; Gursel, D.B.; Lemiere, S.; Reimers, D.; Cuevas, P.; Carceller, F.; Di Santo, J.P.; Bikfalvi, A. Inhibition of fibroblast growth factor/fibroblast growth factor receptor activity in glioma cells impedes tumor growth by both angiogenesis-dependent and -independent mechanisms. Cancer Res. 2001, 61, 1717–1726.
- Jiang, Z.S.; Padua, R.R.; Ju, H.; Doble, B.W.; Jin, Y.; Hao, J.; Cattini, P.A.; Dixon, I.M.; Kardami, E. Acute protection of ischemic heart by FGF-2: Involvement of FGF-2 receptors and protein kinase C. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1071–H1080.
- Jiang, Z.S.; Srisakuldee, W.; Soulet, F.; Bouche, G.; Kardami, E. Non-angiogenic FGF-2 protects the ischemic heart from injury, in the presence or absence of reperfusion. Cardiovasc. Res. 2004, 62, 154–166.
- Pemberton, C.J.; Raudsepp, S.D.; Yandle, T.G.; Cameron, V.A.; Richards, A.M. Plasma cardiotrophin-1 is elevated in human hypertension and stimulated by ventricular stretch. Cardiovasc. Res. 2005, 68, 109–117.
- Pan, J.; Fukuda, K.; Kodama, H.; Sano, M.; Takahashi, T.; Makino, S.; Kato, T.; Manabe, T.; Hori, S.; Ogawa, S. Involvement of gp130-mediated signaling in pressure overload-induced activation of the JAK/STAT pathway in rodent heart. Heart Vessel. 1998, 13, 199–208.
- Feng, Y.; Ye, D.; Wang, Z.; Pan, H.; Lu, X.; Wang, M.; Xu, Y.; Yu, J.; Zhang, J.; Zhao, M.; et al. The Role of Interleukin-6 Family Members in Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 9, 818890.
- Freed, D.H.; Moon, M.C.; Borowiec, A.M.; Jones, S.C.; Zahradka, P.; Dixon, I.M. Cardiotrophin-1: Expression in experimental myocardial infarction and potential role in post-MI wound healing. Mol. Cell. Biochem. 2003, 254, 247–256.
- Guseh, J.S.; Rosenzweig, A. Size matters: Finding growth pathways that protect the heart. Cell Res. 2017, 27, 1187–1188.
- Sano, M.; Fukuda, K.; Kodama, H.; Pan, J.; Saito, M.; Matsuzaki, J.; Takahashi, T.; Makino, S.; Kato, T.; Ogawa, S. Interleukin-6 family of cytokines mediate angiotensin II-induced cardiac hypertrophy in rodent cardiomyocytes. J. Biol. Chem. 2000, 275, 29717–29723.
- Lunde, I.G.; Aronsen, J.M.; Melleby, A.O.; Strand, M.E.; Skogestad, J.; Bendiksen, B.A.; Ahmed, M.S.; Sjaastad, I.; Attramadal, H.; Carlson, C.R.; et al. Cardiomyocyte-specific overexpression of syndecan-4 in mice results in activation of calcineurin-NFAT signalling and exacerbated cardiac hypertrophy. Mol. Biol. Rep. 2022, 49, 11795–11809.
- Luo, Y.; Jiang, N.; May, H.I.; Luo, X.; Ferdous, A.; Schiattarella, G.G.; Chen, G.; Li, Q.; Li, C.; Rothermel, B.A.; et al. Cooperative Binding of ETS2 and NFAT Links Erk1/2 and Calcineurin Signaling in the Pathogenesis of Cardiac Hypertrophy. Circulation 2021, 144, 34–51.
- Chaklader, M.; Rothermel, B.A. Calcineurin in the heart: New horizons for an old friend. Cell. Signal. 2021, 87, 110134.
- Wilkins, B.J.; De Windt, L.J.; Bueno, O.F.; Braz, J.C.; Glascock, B.J.; Kimball, T.F.; Molkentin, J.D. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol. Cell. Biol. 2002, 22, 7603–7613.
- Han, Y.; Nie, J.; Wang, D.W.; Ni, L. Mechanism of histone deacetylases in cardiac hypertrophy and its therapeutic inhibitors. Front. Cardiovasc. Med. 2022, 9, 931475.
- Zhou, H.; Xia, C.; Yang, Y.; Warusawitharana, H.K.; Liu, X.; Tu, Y. The Prevention Role of Theaflavin-3,3′-digallate in Angiotensin II Induced Pathological Cardiac Hypertrophy via CaN-NFAT Signal Pathway. Nutrients 2022, 14, 1391.
- Haq, S.; Choukroun, G.; Kang, Z.B.; Ranu, H.; Matsui, T.; Rosenzweig, A.; Molkentin, J.D.; Alessandrini, A.; Woodgett, J.; Hajjar, R.; et al. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol. 2000, 151, 117–130.
- Li, J.; Wang, J.; Russell, F.D.; Molenaar, P. Activation of calcineurin in human failing heart ventricle by endothelin-1, angiotensin II and urotensin II. Br. J. Pharm. 2005, 145, 432–440.
- Iemitsu, M.; Miyauchi, T.; Maeda, S.; Sakai, S.; Kobayashi, T.; Fujii, N.; Miyazaki, H.; Matsuda, M.; Yamaguchi, I. Physiological and pathological cardiac hypertrophy induce different molecular phenotypes in the rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R2029–R2036.
- Sangaralingham, S.J.; Kuhn, M.; Cannone, V.; Chen, H.H.; Burnett, J.C. Natriuretic peptide pathways in heart failure: Further therapeutic possibilities. Cardiovasc. Res. 2023, 118, 3416–3433.
- Yoshimura, M.; Yasue, H.; Ogawa, H. Pathophysiological significance and clinical application of ANP and BNP in patients with heart failure. Can. J. Physiol. Pharm. 2001, 79, 730–735.
- Dunn, M.E.; Manfredi, T.G.; Agostinucci, K.; Engle, S.K.; Powe, J.; King, N.M.; Rodriguez, L.A.; Gropp, K.E.; Gallacher, M.; Vetter, F.J.; et al. Serum Natriuretic Peptides as Differential Biomarkers Allowing for the Distinction between Physiologic and Pathologic Left Ventricular Hypertrophy. Toxicol. Pathol. 2017, 45, 344–352.
- Engle, S.K.; Watson, D.E. Natriuretic Peptides as Cardiovascular Safety Biomarkers in Rats: Comparison With Blood Pressure, Heart Rate, and Heart Weight. Toxicol. Sci. 2016, 149, 458–472.
- Wong, P.C.; Guo, J.; Zhang, A. The renal and cardiovascular effects of natriuretic peptides. Adv. Physiol. Educ. 2017, 41, 179–185.
- de Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981, 28, 89–94.
- Gorbe, A.; Giricz, Z.; Szunyog, A.; Csont, T.; Burley, D.S.; Baxter, G.F.; Ferdinandy, P. Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Res. Cardiol. 2010, 105, 643–650.
- Elesgaray, R.; Caniffi, C.; Ierace, D.R.; Jaime, M.F.; Fellet, A.; Arranz, C.; Costa, M.A. Signaling cascade that mediates endothelial nitric oxide synthase activation induced by atrial natriuretic peptide. Regul. Pept. 2008, 151, 130–134.
- Cooling, M.; Hunter, P.; Crampin, E.J. Modeling hypertrophic IP3 transients in the cardiac myocyte. Biophys. J. 2007, 93, 3421–3433.
- Kraeutler, M.J.; Soltis, A.R.; Saucerman, J.J. Modeling cardiac beta-adrenergic signaling with normalized-Hill differential equations: Comparison with a biochemical model. BMC Syst. Biol. 2010, 4, 157.
- Ryall, K.A.; Holland, D.O.; Delaney, K.A.; Kraeutler, M.J.; Parker, A.J.; Saucerman, J.J. Network reconstruction and systems analysis of cardiac myocyte hypertrophy signaling. J. Biol. Chem. 2012, 287, 42259–42268.
- Zeigler, A.C.; Richardson, W.J.; Holmes, J.W.; Saucerman, J.J. A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. J. Mol. Cell. Cardiol. 2016, 94, 72–81.
- Zeigler, A.C.; Nelson, A.R.; Chandrabhatla, A.S.; Brazhkina, O.; Holmes, J.W.; Saucerman, J.J. Computational model predicts paracrine and intracellular drivers of fibroblast phenotype after myocardial infarction. Matrix Biol. J. Int. Soc. Matrix Biol. 2020, 91–92, 136–151.
- Zeigler, A.C.; Chandrabhatla, A.S.; Christiansen, S.L.; Nelson, A.R.; Holmes, J.W.; Saucerman, J.J. Network model-based screen for FDA-approved drugs affecting cardiac fibrosis. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 377–388.
- Lu, H.I.; Tong, M.S.; Chen, K.H.; Lee, F.Y.; Chiang, J.Y.; Chung, S.Y.; Sung, P.H.; Yip, H.K. Entresto therapy effectively protects heart and lung against transverse aortic constriction induced cardiopulmonary syndrome injury in rat. Am. J. Transl. Res. 2018, 10, 2290–2305.
- Burke, R.M.; Lighthouse, J.K.; Mickelsen, D.M.; Small, E.M. Sacubitril/Valsartan Decreases Cardiac Fibrosis in Left Ventricle Pressure Overload by Restoring PKG Signaling in Cardiac Fibroblasts. Circ. Heart Fail. 2019, 12, e005565.
- Cunningham, J.W.; Claggett, B.L.; O’Meara, E.; Prescott, M.F.; Pfeffer, M.A.; Shah, S.J.; Redfield, M.M.; Zannad, F.; Chiang, L.M.; Rizkala, A.R.; et al. Effect of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients With HFpEF. J. Am. Coll. Cardiol. 2020, 76, 503–514.

