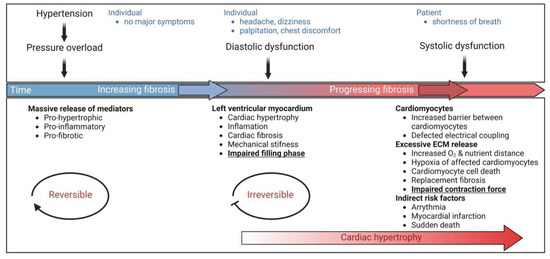
Pathological cardiac hypertrophy is a key risk factor for the development of heart failure and predisposes individuals to cardiac arrhythmia and sudden death. While physiological cardiac hypertrophy is adaptive, hypertrophy resulting from conditions comprising hypertension, aortic stenosis, or genetic mutations, such as hypertrophic cardiomyopathy, is maladaptive. Prolonged cardiovascular stress causes cardiomyocytes and non-myocardial cells to enter an activated state releasing numerous pro-hypertrophic, pro-fibrotic, and pro-inflammatory mediators such as vasoactive hormones, growth factors, and cytokines, i.e., commencing signaling events that collectively cause cardiac hypertrophy. Fibrotic remodeling is mediated by cardiac fibroblasts as the central players, but also endothelial cells and resident and infiltrating immune cells enhance these processes. Many of these hypertrophic mediators are now being integrated into computational models that provide system-level insights and will help to translate our knowledge into new pharmacological targets.
- cardiac hypertrophy
- cardiomyocytes
- heart failure
- myocardial microenvironment
1. General Introduction
2. An Interplay of Different Cells in Hypertrophic Remodeling
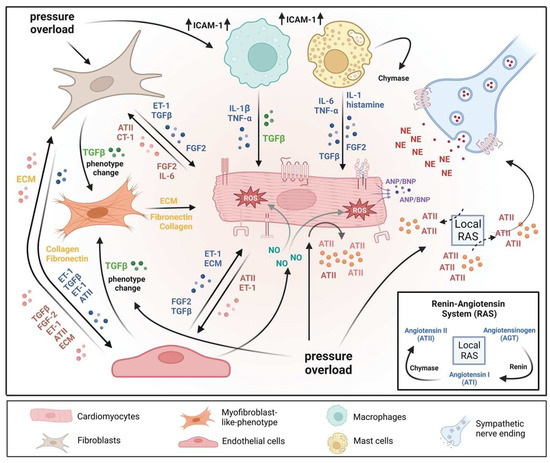
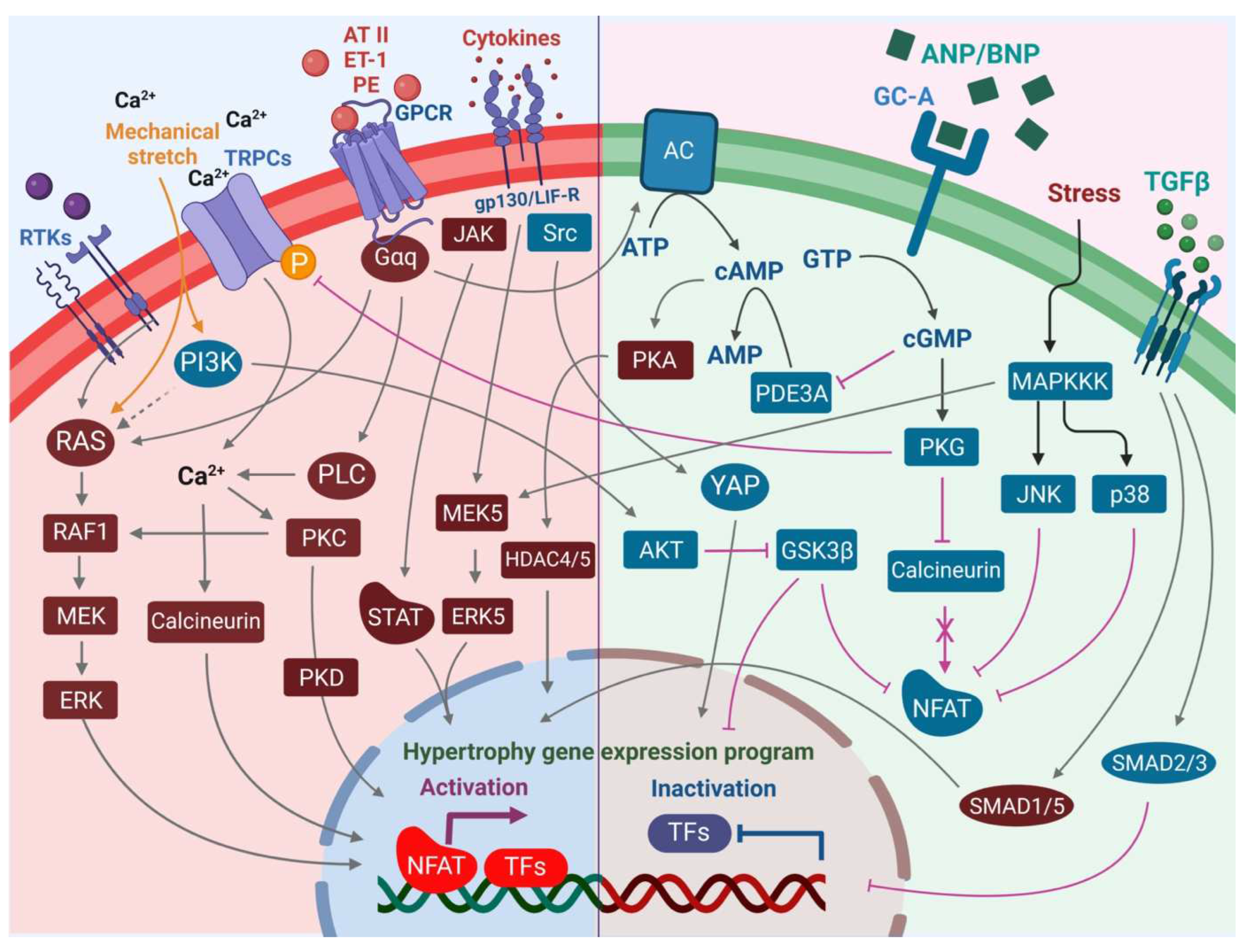
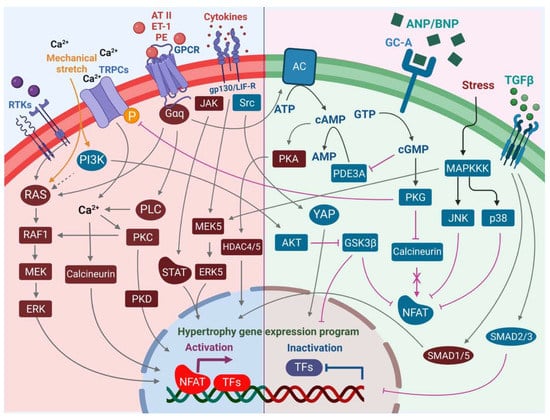
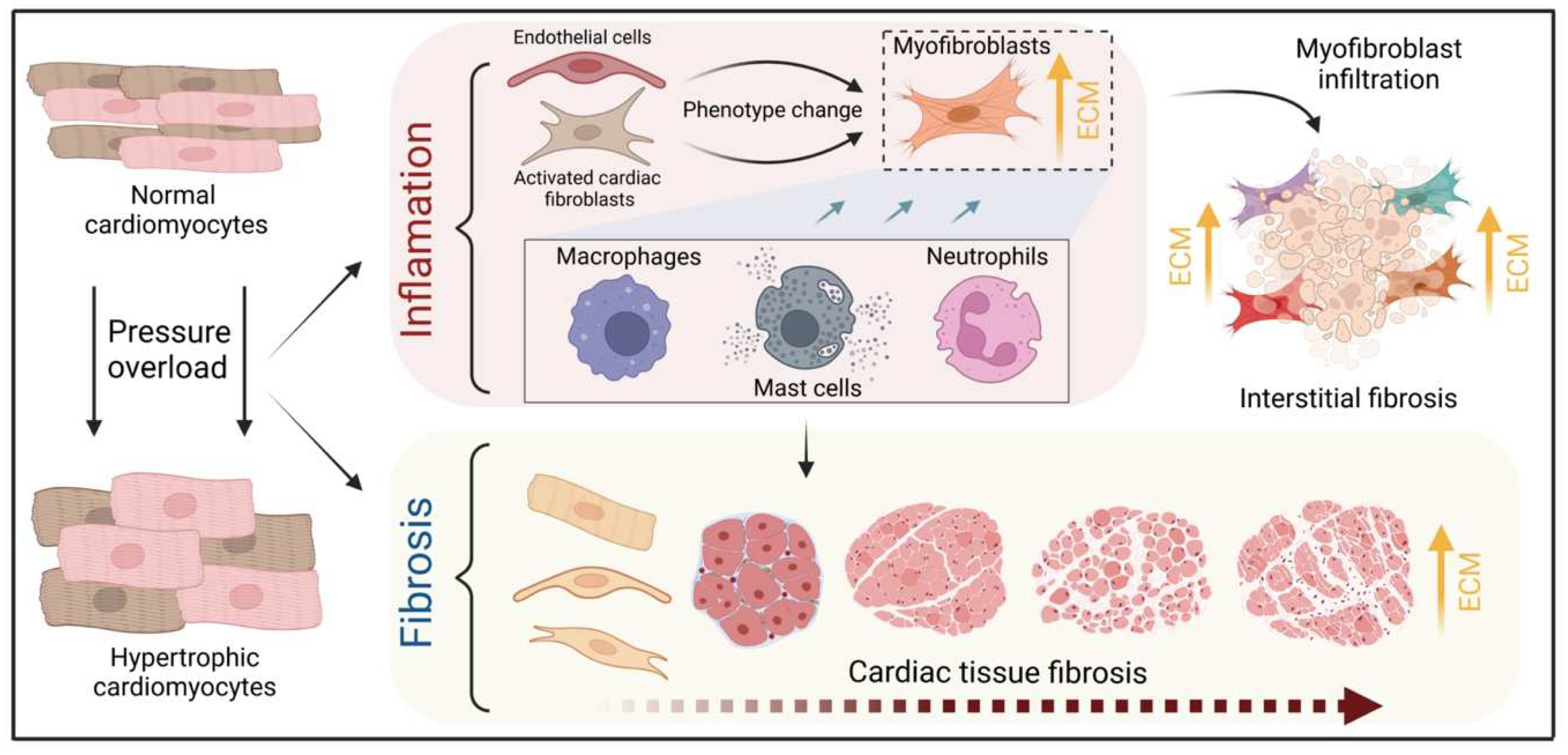
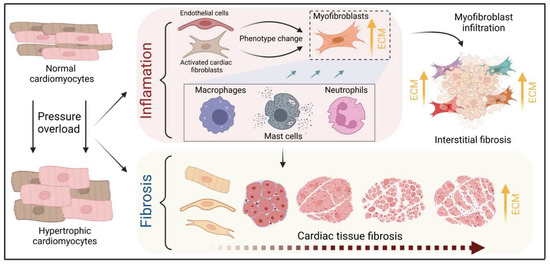
The heart consists of various cell types, including myocytes, endothelial cells, fibroblasts, vascular smooth muscle cells, sympathetic neurons, and immune cells, which collectively account for a synchronized cardiac function [10][11][23,24]. However, it has been shown that owing to their enormous size, cardiomyocytes in particular account for the majority of heart mass, increase in size and reprogram transcription in the process of cardiac hypertrophy [2][12][2,25]. Communications between cardiomyocytes and non-myocytes lead to the secretion of bioactive mediators, which operate in an autocrine and paracrine manner. This is followed by microenvironmental stimulation of different cell types and the activation of various signaling pathways within the cells (Figure 1 and Figure 2) [13][14][26,27]. Altogether these complex processes result in cardiomyocyte hypertrophy, fibroblast hyperplasia, interstitial tissue composition changes, and remodeling of the ventricular chambers [15][28].2.1. Fibroblast Remodeling
Pressure overload triggers resident cardiac fibroblasts originating from the epicardium and endocardium to undergo rapid expansion and activation, rather than previously reported hematopoietic precursor-derived fibroblasts or endothelial-to-mesenchymal transition (EndMT) as a contributing source (Figure 1 and Figure 3) [16][17][29,30]. Despite this, the exact origins of cardiac fibroblasts as well as the delineation of their characteristics and plasticity remain a field of current investigation and controversy [18][31]. Like cardiomyocytes, fibroblasts respond to external stress stimuli, but in a slightly different manner. Mechanical stress promotes fibroblast differentiation to a myofibroblast-like phenotype (Figure 1 and Figure 3) [19][20][32,33], which has been shown to develop from tissue-derived fibroblasts rather than endothelial or smooth muscle cells [17][30].2.2. Endothelial Cell Activation
In response to pressure overload, cardiac endothelial cells, similar to cardiac fibroblasts, are capable of changing their phenotype (Figure 1). It has been reported that endothelial cells can undergo an EndMT, differentiate into myofibroblast-like cells, and thereby contribute to cardiac fibrosis [21][38]. Others outlined that EndMT recruits circulating hematopoietic progenitors to the heart thereby generating significant numbers of cardiac fibroblasts (reviewed in [22][39]) but also their origin from tissue-resident fibroblasts is being discussed [16][17][29,30]. Altogether, left ventricular myocardial tissue of end-stage cardiac failure patients revealed dramatically increased expression levels of EndMT-related genes [23][40], indicating the need for further investigation to clarify the exact contribution of EndMT.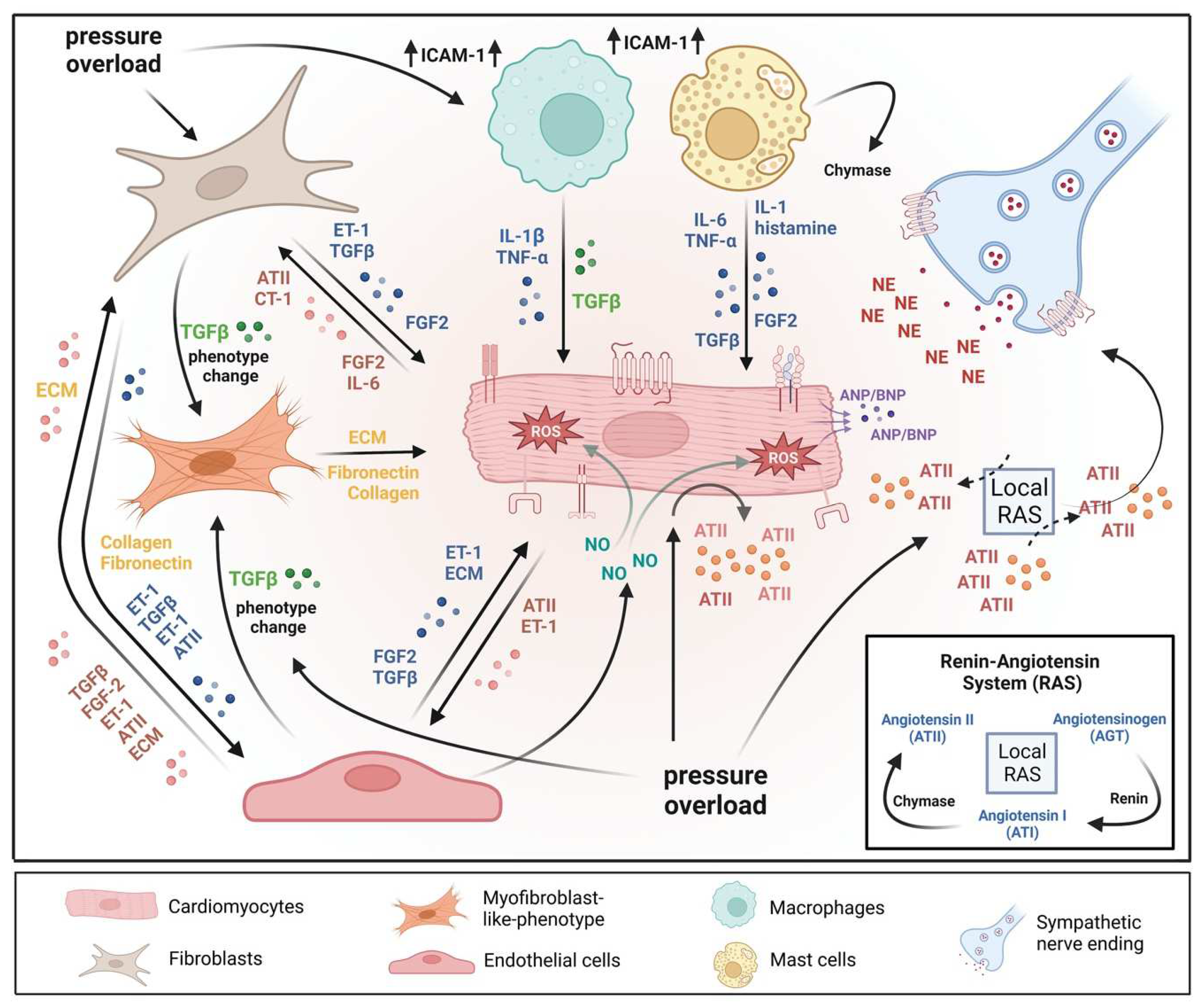
3. The Role of Immune Cells in Cardiac Hypertrophy
3.1. Cardiac Mast Cells
3.2. Monocytes & Macrophages
Healthy and injured cardiac tissues possess heterogeneous populations of macrophages, in both humans and mice (Figure 1) [45][129]. Most macrophages within the heart are established embryonically from the yolk sac and fetal liver progenitors, similar to tissue macrophages of the liver or brain. Local proliferation in contrast to monocyte recruitment serves to maintain resident macrophage subsets [46][47][130,131]. In the absence of disease, self-renewal serves to maintain local tissue macrophage populations [48][132]. Despite this, in response to pressure overload or ischemic injuries, the majority of macrophages are derived from the recruitment and differentiation of blood monocytes [49][133]. Cardiac macrophages are key effector cells mediating tissue remodeling and fibrosis (Figure 3) [50][134]. The initial and significant event for vascular lesion formation results from inflammatory cytokine- and growth factor-producing migrating macrophages (Figure 1) [51][135]. The accumulation of macrophages has been found in the perivascular space, where they co-localize with fibroblasts collectively producing collagen during cardiac hypertrophy (Figure 3) [52][53][136,137].3.3. Neutrophils
Under normal reparative conditions, neutrophil granulocytes are recruited to areas of acute inflammation, where they perform functions such as the clearance of dead cells and matrix debris (Figure 3) [54][55][141,142]. As key components of the inflammatory response, neutrophils also act on the recruitment, activation, and programming of antigen-presenting cells (APCs). Specifically, they attract monocytes and dendritic cells (DCs) by generating chemotactic signals, thereby influencing the differentiation of macrophages into a predominantly pro- or anti-inflammatory state [56][57][58][143,144,145]. Because neutrophil granulocytes are one of the most important cellular components of the body for the destruction of microorganisms, there is also the possibility that these cells damage host cells and tissues [59][146]. Neutrophils have been described to produce cytokines such as TNFα that drive macrophage and dendritic cell differentiation [56][58][60][143,145,155]. Additionally, neutrophilic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase gets activated in response to pressure overload injury [61][156], resulting in the degranulation of neutrophils and thereby release of pro-fibrotic proteases (Figure 3) as well as reactive oxygen species (ROS) [62][157].3.4. Lymphocytes
A growing body of research indicates that systemic inflammation may play a significant pathophysiologic role in the etiology of cardiac disease development, including HCM, and may have an impact on the severity of the phenotypic and clinical outcomes, including heart failure. A high neutrophil-to-lymphocyte ratio (NLR), a marker of oxidative stress damage, has been linked to an increased 5-year risk of sudden cardiac death associated with HCM [63][64][158,159], which supported further the prognostic significance of inflammation. However, in angiotensin II-induced HF models, the absence of B cells led to less hypertrophy and collagen deposition, the preservation of left ventricular function, and, in conjunction with these changes, a decrease in the expression of proinflammatory cytokines and apoptosis in the myocardium [65][161]. Different studies have also reported that activation of NK T cells improved cardiac remodeling events and failure in mice by increasing the expression of cardioprotective cytokines, including IL-10 [66][67][162,163].3.5. Sympathetic Neurons
Sympathetic neurons that innervate the heart and release norepinephrine (NE) also express the endothelin receptor A (ET-A) [68][69][168,169]. ET-resulted in a tremendous NE release in cocultured cardiomyocytes and sympathetic neurons with exaggerated hypertrophy of cardiomyocytes compared to monocultured cardiomyocytes. In contrast, mice lacking the ET-A receptor exclusively in sympathetic neurons showed less adverse structural remodeling, and cardiac dysfunction when exposed to pathological pressure overload [70][170]. Substantial amounts of renin released in the cardiac microenvironment upon cardiac mast cell degranulation [71][65] result in both AT-II formation within striking distance of AT1 receptor-expressing cardiac sympathetic nerve terminals and enhanced NE release (Figure 1) and arrhythmias (Figure 4) [72][73][171,172].