Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael E. Herbig | -- | 3745 | 2023-07-06 15:17:09 | | | |
| 2 | Jason Zhu | Meta information modification | 3745 | 2023-07-07 04:14:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Herbig, M.E.; Evers, D.; Gorissen, S.; Köllmer, M. Characterization of Semi-Solid Formulations. Encyclopedia. Available online: https://encyclopedia.pub/entry/46525 (accessed on 26 July 2026).
Herbig ME, Evers D, Gorissen S, Köllmer M. Characterization of Semi-Solid Formulations. Encyclopedia. Available at: https://encyclopedia.pub/entry/46525. Accessed July 26, 2026.
Herbig, Michael E., Dirk-Heinrich Evers, Sascha Gorissen, Melanie Köllmer. "Characterization of Semi-Solid Formulations" Encyclopedia, https://encyclopedia.pub/entry/46525 (accessed July 26, 2026).
Herbig, M.E., Evers, D., Gorissen, S., & Köllmer, M. (2023, July 06). Characterization of Semi-Solid Formulations. In Encyclopedia. https://encyclopedia.pub/entry/46525
Herbig, Michael E., et al. "Characterization of Semi-Solid Formulations." Encyclopedia. Web. 06 July, 2023.
Copy Citation
Specific aspects of semi-solid dosage forms for topical application include the nature of the barrier to be overcome, aspects of susceptibility to physical and chemical instability, and a greater influence of sensory perception. Advances in understanding the driving forces of skin penetration as well as the design principles and inner structure of formulations, provide a good basis for the more rational design of such dosage forms, which still often follow more traditional design approaches. The characterization of semi-solid dosage forms also requires specific approaches. In particular, rheology, chemical stability considering the specifics of semi-solid vehicles, and in vitro performance testing will be discussed.
skin penetration
chemical stability
rheology
excipients
analytics
1. Introduction
Topical semi-solid formulations, especially when emulsion-based, are often systems of high complexity, requiring sophisticated approaches for the characterization of chemical, physical, and microbial stability. In earlier stages of development, characterization is more focused on decision-making, knowledge-gaining, and risk mitigation, whereas the focus in later stages is on establishing data packages for submission and quality control. Especially in the context of generic drug development, the microscale organization of matter in semi-solids is often described as “microstructure.” Physical characterization may include microscopy, rheology, light scattering and laser diffraction techniques for particle size characterization, and a number of spectroscopic techniques. For comprehensive overviews of microstructure characterization in topicals, the reader can be referred to the book by Langley, Michniak–Kohn and Osborn [1] and the article by Badruddoza et al. [2]. What will be discussed in the following are applications of rheology for formulation characterization, an area that gained importance during the last years and were interesting case studies became available. Approaches to address chemical stability in semi-solid formulations are less described in the literature than aspects of physical stability but are often critical for successful development. Therefore, also this aspect will be discussed. Finally, a brief overview of in vitro methods to investigate release, penetration, and permeation form semi-solid formulations will be given.
2. Rheological Characterization of Semi-Solids
The rheological characterization of semi-solid formulations provides an important source of information on various aspects relevant to the systematic development and characterization of semi-solid formulations. Rheological characteristics can be used to describe several technological processes, such as pipe flow, properties during stirring or spraying, product removal from the packaging or how the product can be applied on the skin. Semi-solid formulations are typically structured liquids that follow non-Newtonian flow behaviors. This means that their viscosity is dependent on the shear applied. Most semi-solid dosage forms show a shear-thinning flow behavior. Whereas the viscosity at moderate and elevated shear rates is easier to determine, viscosity values at very low shear rats of 0.001 1/s or below can only be measured by modern high-performance rheometers. Values at such low shear rates are often also referred to as ‘zero shear viscosity.’ Whenever the viscosity of a formulation at rest needs to be described, the zero-shear viscosity needs to be considered. This refers to the phenomena of sedimentation or creaming in emulsions and suspensions. According to the Stokes equation, the speed of creaming or sedimentation is inversely proportional to the dynamic viscosity [3]. According to the Stokes–Einstein equation, also the diffusion coefficient is inversely proportional to the viscosity. Whenever systems at rest are described, the zero-shear viscosity needs to be considered. In Figure 1, the viscosity curves of two creams thickened by different polymers are presented in double-logarithmic presentation (M. Köllmer, unpublished data). Whereas the viscosity at high shear rates and, therefore, the spreadability is almost identical, cream 1 has a five-fold higher zero-shear viscosity which makes it more stable against creaming or sedimentation.
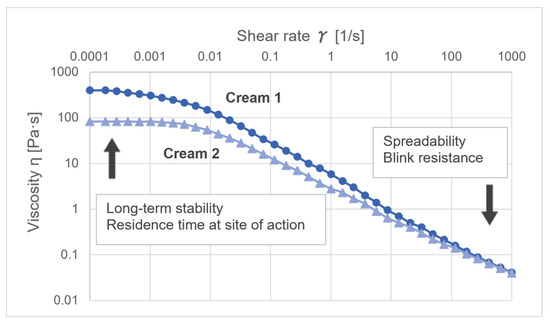
Figure 1. Differences in shear-thinning behavior of two creams. While both creams show comparable viscosity at higher shear rates, cream 1 (dots) has a 5-times higher zero-shear viscosity than cream 2 (triangles).
The viscoelastic properties of semi-solid formulations can be obtained using oscillatory tests such as the amplitude sweep or the frequency sweep test. The most relevant parameters for semi-solid formulation development include the loss modulus (symbol G″, unit Pa) that describes the viscous content of a formulation, which is a measure of the lost deformation energy during the test. This energy is used for the alteration of the sample structure and is delivered to the surrounding area. Ideal viscous substances have the same loss moduli before and after stress application. The storage modulus (symbol G′, unit Pa) describes the elastic content of a formulation. It is a measure of the stored deformation energy during the test. Ideal elastic substances have the same storage moduli before and after stress application. The phase angle (symbol δ, unit °) describes the ratio between viscous (liquid-like) and elastic (solid-like) components. A value of 0° describes an ideally elastic material (e.g., steel), and a value of 90° describes a liquid such as water. Materials with phase angles < 45° do not flow when at rest. For comparable materials, a lower phase angle is an indication of a more pronounced internal structure. The flow point (symbol τFP, unit Pa) is the shear stress value at the intersection of the curves of G″ and G′. If the flow point is exceeded, the viscous portion dominates over the elastic part of a sample, and it flows. A high flow point often is a measure of good structural stability.
Rheological investigations can contribute to the characterization, prediction and optimization aspects of formulation aging and stability [4][5]. Changes in the formulation microstructure over time or after exposure to different temperatures can often be detected more sensitively in the rheological profile than in macroscopic or microscopic examination. In particular, the rheological swing test—also called the temperature cycling test—introduced by Brummer [5], can provide valuable contributions to the prediction of the long-term physical stability of emulsion systems. If the loss factor tanδ (or, alternatively, the storage or complex modulus) remains constant after several temperature cycles, this is a good indicator of long-term physical stability. Formulations that show significant changes in these parameters, in contrast, are prone to physical instability.
Furthermore, inter-batch variability due to different excipient batches, altered manufacturing conditions, or during storage can be determined. The similarity of the microstructure of a generic cream to an originator product can be assessed by rheological equivalence testing. The EMA Draft guideline on the quality and equivalence of topical products from 2019 provides specific guidance on equivalence testing for topical products in lieu of clinical equivalence trials [6].
Rheological investigations have also been used to investigate correlations of viscosity with release and permeation from semi-solid formulations. Whereas, as expected from the Stokes-Einstein equation, a negative correlation of viscosity with the release rate could be demonstrated, no clear correlation with skin permeation or penetration could be observed [7][8]. The latter is not surprising, as the rate-limiting step for penetration into the skin is rather the diffusion through the stratum corneum than the diffusion within the formulations. Furthermore, rheology has proven to be useful in predicting certain sensorial properties [9][10] and process parameters. In a recent study, it has been demonstrated that a thixotropic emulsion gel-based nasal spray could be optimized by means of rheology for device compatibility and improvement of nasal residence time [11].
In the opinion of the authors, a comprehensive rheological analysis should be part of the formulation development and optimization work for semi-solid formulations. However, it is not recommended to perform the same set of tests for each formulation but to select the rheological test based on an analysis of which parameters are critical to the performance of a particular formulation. For example, for a pourable lotion, zero-shear viscosity may be critical because it correlates with stability against creaming or sedimentation if it contains suspended particles. In contrast, for a petrolatum-based ointment, it may be important that the viscosity at higher shear rates remains below a certain limit to ensure acceptable spreadability on the skin.
2. Chemical Analytics of Semi-Solids
The development of analytical methods is equally important as formulation development. Successful product development consists of the close interlinking and collaboration of formulation development and analytical method development. Only with efficient and reliable analytical methods can product quality be ensured, optimized and controlled. In other words, the certainty of having a good product can only be as high as the quality of the methods used to analyze it.
In many cases, analytical method development for semi-solid formulations is more challenging, for example, for solid dosage forms. One reason is the typically unfavorable drug-to-matrix ratio. A tablet often consists of around 10–50% active ingredient, whereas for semi-solid formulations, this is often between 0.1% and 1%. The level at which degradation products need to be determined can be calculated from their concentration in the formulation and the (anticipated) maximum daily dose (MDD) according to ICH Q3B [12]. An illustration of the levels of the reporting, identification, and qualification thresholds for a 0.1% formulation dependent on the MDD is shown for a formulation with 0.1% API in Figure 2. It can be seen that impurities often need to be determined at a 0.1% level of the active ingredient. For a product with 0.1% API, this results in a quantification limit of 0.0001% for the impurity. Apart from the need for well-developed methods and sensitive equipment, this may also need sophisticated extraction procedures from the formulation matrix. The task of the formulator is to develop a stable formulation that does not easily disintegrate upon thermal or mechanical stress, whereas the analytical scientist has to extract both the API and its impurities quantitatively from the same formulation matrix. Creams and ointments are often composed of excipients with different polarity, solubility, and melting points. A good understanding of chemistry, formulation design, excipient properties and often creative problem-solving skills are necessary to develop efficient and robust extraction procedures. Another specific challenge of semi-solid products lies in the fact that the API, as opposed to solid dosage forms, is typically present in a dissolved state and can, therefore, undergo chemical reactions more easily. APIs can undergo basic or acidic hydrolysis, various types of oxidation, isomerization, transesterification or migration into or through different types of packaging material. The same also applies to functional excipients such as antioxidants or preservatives whose stability also needs to be monitored. Furthermore, the compatibility of the API with excipients needs to be investigated. As pure excipients may behave differently from the excipient, which is formulated in a complex matrix, two approaches are possible: The stability is first investigated at the level of the formulation, and if an indication for incompatibility with excipients exists, a compatibility screening with potentially responsible excipients is performed. Alternatively, a comprehensive compatibility screening is performed prior to the start of the formulation development. This, however, is associated with the risk of discarding excipient options, which are only problematic when used as pure excipients but not as part of a complex formulation.
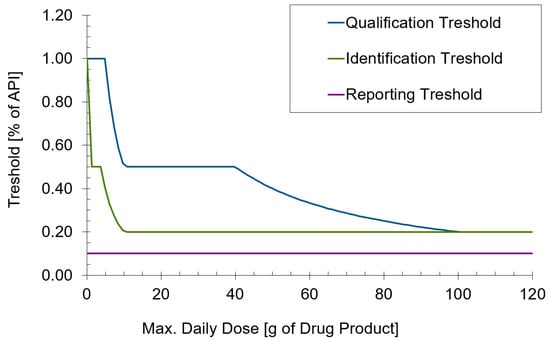
Figure 2. Illustration of the dependency of qualification, identification, and reporting thresholds for impurities dependent on the maximum daily dose (MMD) according to ICH Q3B [12].
Method development should start with stress tests, which are also referred to as forced degradation studies, to obtain information on the stability and degradation behavior of the API when exposed to different stress conditions. Stress tests are mandatory as part of the stability studies for the registration application of new drug substances or drug products as described in the ICH Q 1 A (R2) “Stability Testing of new Drug Substances and Products” [13]. The guideline states that “Stress testing of the drug substance can help identify the likely degradation products, which can in turn help establish the degradation pathways and the intrinsic stability of the molecule and validate the stability-indicating power of the analytical procedures used. The nature of the stress testing will depend on the individual drug substance and the type of drug product involved.” Thermal, oxidative, photolytic and hydrolytic (acidic and basic) stress conditions are required. It is important to be aware that a decrease in the API peak in the chromatographic analysis may not necessarily be connected to a corresponding increase in peaks of degradation products. The earlier a mass balance of the API and its degradation products can be established, the better. For registration, this is mandatory. It is even possible that despite substantial API degradation, there are no peaks of degradation products visible in the chromatograms in case their chromophore is destroyed. Therefore, it is recommendable that detection at multiple wavelengths and/or mass-spectrometric detection is used for stress test studies [14]. The exact experimental conditions are not defined, but potential approaches have been described in the literature [15].
Although this ICH guideline is not binding for early explorative stages of development, it provides useful general guidance. The value generated from properly designed stress tests is in the identification of degradation mechanisms such as hydrolysis, oxidation, thermolysis or photolysis and the establishment of preliminary hypotheses for degradation pathways and possible structures of API-related degradation products (especially if in addition to UV detection, molecular masses are also recorded, e.g., by routinely using an additional single-MS detector). This provides an important basis and good understanding of the API molecule, the identification of options to optimize stability in formulations, and for the development of stability-indicating analytical methods and, ultimately, robust drug products. It may be important to adapt the stress conditions to the API, e.g., to avoid ‘over stress’ resulting in the dominance of secondary degradation products. In such a case, important information would be lost.
Although not a formal requirement in exploratory development, from a risk mitigation point of view, it is important to know if an analytical method is stability-indicating or not as soon as significant investment decisions are made, such as entering the clinical phase or during the process of due diligence for a product to be licensed. This is especially the case if conclusions should be made based on the assay. Without that knowledge, there is a substantial risk of making false-positive decisions on drug product stability. According to Blessy et al. [15], a stability-indicating method (SIM) is an analytical procedure used to quantitate the decrease in the amount of the active pharmaceutical ingredient (API) in drug products due to degradation. A stability-indicating method accurately measures the changes in active ingredient concentration without interference from other degradation products, impurities and excipients. Often, this is confirmed by investigations on spectral homogeneity of the relevant peak(s) in the chromatogram. However, it should be considered that in topical formulations, often complex excipients are used, which may elute over almost the entire range of retention time, as, for example, demonstrated for polysorbate 20 [16]. Alternatively, or in combination with stability-indicating assay methods, a comprehensive analysis of the degradation products based on a good understanding of the degradation kinetics is very helpful in the risk assessment of prototype formulations.
Chemical stability studies in exploratory development are typically performed in the form of accelerated stability studies. As opposed to ICH stability studies, the focus is not on providing data for registration purposes but on predictive risk-analysis and formulation selection under accelerated conditions, i.e., with substantial time savings as compared to real-time studies. Typically, formulations are stored at various temperatures between 5 °C and 40 °C. If the formulations are stable at these temperatures, also 50 °C or even 60 °C may be considered. However, formulations that show major physical changes, such as phase separation or precipitation, should not be included in the analysis as the chemical degradation behavior may be influenced by these physical changes. In exploratory accelerated studies of semi-solid formulations, typically, hermetic packaging materials (e.g., glass vials with screw caps) are used so that controlled humidity is not relevant for the study.
If the reaction constants generated from the stability studies follow the Arrhenius equation, data from 12 week accelerated stability studies often allow a preliminary shelf-life prediction. If a deviation from the Arrhenius equation is observed, a root cause analysis needs to be performed to evaluate if, for example, physical changes in the formulation or a deviation from the assumed reaction kinetic occurred (e.g., incorrect reaction order or subsequent reaction).
If an API is chemically very stable in a formulation and no relevant degradation occurs even at higher temperatures, it will also not be possible to establish Arrhenius calculations. In such a case, however, it will be possible to still assume that achieving a commercial shelf life will be feasible.
The approach to the development of analytical methods should follow a structured and strategic approach already in the early stages of development. Although not a formal requirement in exploratory development, the concept of defining an analytical target profile (ATP) is recommended. According to USP-NF 〈1220〉 “Analytical Procedure life Cycle” [17], an “ATP is a prospective description of the desired performance of an analytical procedure that is used to measure a quality attribute, and it defines the required quality of the reportable value produced by the procedure, aligned with the quality target product profile (QTPP).” The benefits of establishing a preliminary ATP already in the early stages of development are that an explicit reflection on the design goals for a new analytical procedure takes place. The efforts of establishing certain performance attributes of analytical methods can be weighed against the risks of making incorrect decisions when skipping such efforts. Furthermore, an ATP is a useful tool for alignment between product development and other stakeholders or sponsors. In USP-NF 〈1220〉, the ATP is part of the analytical procedure life cycle, which is defined in three stages. Many of the elements of stage 1 are already the best investigated in exploratory development, such as “understanding gained through knowledge gathering, systematic procedure development experiments, and risk assessments and associated lab experiments.” It may be difficult, risky, and require more resources if these elements are investigated only retrospectively. An advantage of the ATP is that the performance criteria of an analytical method are defined rather than a concrete analytical procedure. As a consequence, changes in the concrete procedure of such a method during its life cycle can be approved by the authorities much more easily.
The strategies for stability testing and optimization are highly API and formulation dependent. Apart from the guiding principles outlined above, it is essential not just to follow a general protocol but to work with the generated data and to anticipate potential issues based on the properties of API, formulation, excipients and primary packaging. Compatibility with single excipients may not be predictive of the compatibility at the level of the formulation as dilution or distribution between phases or into the oil-water interface may influence stability. Furthermore, APIs may not react directly with excipients but only with degradation products of excipients or with impurities of excipients, which may be present in higher or lower concentrations, dependent on the batch or age and storage conditions of the excipients. Furthermore, depending on both the properties of the API and the formulation, APIs may migrate into or through the primary packaging or undergo degradation processes catalyzed by certain components of the packaging material.
3. In Vitro Performance Testing
The classical method for investigating formulation performance is the Franz diffusion cells method established in 1975 [18]. Originally, they were performed as in vitro permeation studies by using skin as a membrane, an experiment established as in vitro permeation testing (IVPT). Alternatively, also filter membranes that only provide a separation of the formulation from the acceptor compartment without providing a relevant barrier for diffusion can be used in an experiment called in vitro release testing (IVRT). Except for the similarities in testing instrumentation, the two methods are not practically comparable as the differences in goals, purposes, and techniques far outweigh the similarities, as summarized in Table 1. Whenever the release of the API from the matrix is a rate-limiting or critical formulation attribute, IVRT may be a suitable tool for performance characterization. In most cases, however, it is rather a quality control tool. The primary readout parameter for IVPT studies is the flux, which is an important parameter for the initial assessment of systemic exposure, which is relevant for safety assessment or transdermal drug delivery. It may also be used as a surrogate for penetration into the skin.
Table 1. Differences between in vitro release testing (IVRT) and in vitro permeation testing (IVPT).
| Parameter | IVRT | IVPT |
|---|---|---|
| Investigated process | Release | Permeation |
| Membrane used | Synthetic filter membrane | (human) skin |
| Donor chamber exposure | Occluded | Often unoccluded |
| Dosing | Infinite dose | Often (semi)finite dose |
| Readout parameters | Flux profile (Jmax, etc.) | Release rate (slope) |
| Receptor cell media | Non-physiological media acceptable | Physiological media preferred |
| Typcial detection range | µg/mL range | ng/mL range |
| Contact of product with acceptor | Product-media interface | Product stays “dry“ |
| Variability in membrane | Relatively consistent in quality | Donor variability |
| in vitro/in vivo correlation | Not designed to correlate with in vivo | IVIV correlation expected |
| Overall purpose | Assessment of quality | Assessment of performance/safety |
The skin membrane used in the IVPT experiment can also be extracted, and the concentration in different skin layers can be determined. To separate the skin layers, either slicing by a microtome [19] or heat separation of the epidermis and dermis can be used [20]. IVPT and IVRT are described in regulatory guidelines and are important tools for the bioequivalence assessment of topical generic drugs. Comprehensive studies of their use in this context have been published [21][22]. In early development, especially for compound and formulation selection, alternative models can be used. The “Hamburg model of skin penetration” [23] allows the determination of cutaneous biodistribution in viable pig ear skin. For a diverse set of compounds and formulations, it has shown excellent correlation to viable human skin. As the skin remains metabolically viable throughout the experiment, it also offers the advantage that the metabolism of APIs in the skin can be monitored. Apart from the fact that the viability of the skin was not considered, a similar model has been described by Quartier et al. [24]. Dermal open-flow microperfusion and dermal microdialysis, which are more frequently performed as in vivo investigations, have also been demonstrated as potential tools for ex vivo studies on the pharmacokinetics of topically applied drugs [25]. Although the site of action for most topically applied drugs is in the epidermis or dermis, also stratum corneum sampling by tape stripping may be used as a surrogate model [26][27][28]. In recent years, also spectroscopic techniques, mainly by confocal Raman microscopy, have been used for in vitro skin penetration assessment with promising results [29][30][31][32].
Overall, studying drug release, penetration, or permeation by in vitro models is an important part of the early development of topical semi-solid dosage forms. It is important to understand the potential and limitations of the respective models with regard to the purpose of the investigations. Aspects like the site of action within the skin, detectability, potential metabolism of the API, infinite or finite dosing, and transformation of the vehicle need to be considered.
References
- Langley, N.; Michniak-Kohn, B.; Osborne, D.W. (Eds.) The Role of Microstructure in Topical Drug Product Development; Springer International Publishing: Cham, Switzerland, 2019.
- Badruddoza, A.Z.M.; Yeoh, T.; Shah, J.C.; Walsh, T. Assessing and Predicting Physical Stability of Emulsion-Based Topical Semisolid Products: A Review. J. Pharm. Sci. 2023, 112, 1772–1793.
- Scherphof, G.L.; Fahr, A. Voigt’s Pharmaceutical Technology; Wiley: Hoboken, NJ, USA, 2018.
- Rawat, A.; Gupta, S.S.; Kalluri, H.; Lowenborg, M.; Bhatia, K.; Warner, K. Rheological Characterization in the Development of Topical Drug Products. In The Role of Microstructure in Topical Drug Product Development; Langley, N., Michniak-Kohn, B., Osborne, D.W., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 3–45.
- Brummer, R. Rheology Essentials of Cosmetic and Food Emulsions; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2016.
- CHMP/QWP/708282/2018; Draft Guideline on Quality and Equivalence of Topical Products. European Medicines Agency: Amsterdam, The Netherlands, 2018.
- Dong, L.; Liu, C.; Cun, D.; Fang, L. The effect of rheological behavior and microstructure of the emulgels on the release and permeation profiles of Terpinen-4-ol. Eur. J. Pharm. Sci. 2015, 78, 140–150.
- Binder, L.; Mazál, J.; Petz, R.; Klang, V.; Valenta, C. The role of viscosity on skin penetration from cellulose ether-based hydrogels. Ski. Res. Technol. 2019, 25, 725–734.
- Calixto, L.S.; Infante, V.H.P.; Campos, P.M.B.G.M. Design and Characterization of Topical Formulations: Correlations Between Instrumental and Sensorial Measurements. AAPS PharmSciTech 2018, 19, 1512–1519.
- Moravkova, T.; Filip, P. Relation between sensory analysis and rheology of body lotions. Int. J. Cosmet. Sci. 2016, 38, 558–566.
- Sailer, M.M.; Köllmer, M.; Masson, B.; Fais, F.; Hohenfeld, I.P.; Herbig, M.E.; Koitschev, A.K.; Becker, S. Nasal residence time and rheological properties of a new bentonite-based thixotropic gel emulsion nasal spray—AM-301. Drug Dev. Ind. Pharm. 2023, 49, 103–114.
- European Medicines Agency. CHMP/ICH, Q 3 B (R2) Impurities in New Drug Products; European Medicines Agency: Amsterdam, The Netherlands, 2006.
- European Medicines Agency. CHMP/ICH, Q 1 A (R2) Stability Testing of new Drug Substances and Products; European Medicines Agency: Amsterdam, The Netherlands, 2003.
- Bakshi, M.; Singh, S. Development of validated stability-indicating assay methods—Critical review. J. Pharm. Biomed. Anal. 2002, 28, 1011–1040.
- Blessy, M.; Patel, R.D.; Prajapati, P.N.; Agrawal, Y.K. Development of forced degradation and stability indicating studies of drugs—A review. J. Pharm. Anal. 2014, 4, 159–165.
- Evers, D.-H.; Schultz-Fademrecht, T.; Garidel, P.; Buske, J. Development and validation of a selective marker-based quantification of polysorbate 20 in biopharmaceutical formulations using UPLC QDa detection. J. Chromatogr. B 2020, 1157, 122287.
- USP-NF. 1220 Analytical Procedure Life Cycle; USP-NF: North Bethesda, ML, USA, 2022.
- Franz, T.J. Percutaneous absorption on the relevance of in vitro data. J. Investig. Dermatol. 1975, 64, 190–195.
- Lunter, D.; Daniels, R. In Vitro skin permeation and penetration of nonivamide from novel film-forming emulsions. Ski. Pharmacol. Physiol. 2013, 26, 139–146.
- Carrer, V.; Alonso, C.; Oliver, M.A.; Coderch, L. In Vitro penetration through the skin layers of topically applied glucocorticoids. Drug Test. Anal. 2018, 10, 1528–1535.
- Raney, S.G.; Ghosh, P.; Ramezanli, T.; Lehman, P.A.; Franz, T.J. Cutaneous Pharmacokinetic Approaches to Compare Bioavailability and/or Bioequivalence for Topical Drug Products. Dermatol. Clin. 2022, 40, 319–332.
- Miranda, M.; Veloso, C.; Brown, M.; Pais, A.A.C.C.; Cardoso, C.; Vitorino, C. Topical bioequivalence: Experimental and regulatory considerations following formulation complexity. Int. J. Pharm. 2022, 620, 121705.
- Herbig, M.E.; Houdek, P.; Gorissen, S.; Zorn-Kruppa, M.; Wladykowski, E.; Volksdorf, T.; Grzybowski, S.; Kolios, G.; Willers, C.; Mallwitz, H.; et al. A custom tailored model to investigate skin penetration in porcine skin and its comparison with human skin. Eur. J. Pharm. Biopharm. 2015, 95, 99–109.
- Quartier, J.; Capony, N.; Lapteva, M.; Kalia, Y.N. Cutaneous Biodistribution: A High-Resolution Methodology to Assess Bioequivalence in Topical Skin Delivery. Pharmaceutics 2019, 11, 484.
- Holmgaard, R.; Benfeldt, E.; Nielsen, J.B.; Gatschelhofer, C.; Sorensen, J.A.; Höfferer, C.; Bodenlenz, M.; Pieber, T.R.; Sinner, F. Comparison of open-flow microperfusion and microdialysis methodologies when sampling topically applied fentanyl and benzoic acid in human dermis ex vivo. Pharm. Res. 2012, 29, 1808–1820.
- Mohammed, D.; Matts, P.J.; Hadgraft, J.; Lane, M.E. Depth profiling of stratum corneum biophysical and molecular properties. Br. J. Dermatol. 2011, 164, 957–965.
- N’Dri-Stempfer, B.; Navidi, W.C.; Guy, R.H.; Bunge, A.L. Improved bioequivalence assessment of topical dermatological drug products using dermatopharmacokinetics. Pharm. Res. 2009, 26, 316–328.
- Pensado, A.; Chiu, W.S.; Cordery, S.F.; Rantou, E.; Bunge, A.L.; Delgado-Charro, M.B.; Guy, R.H. Stratum Corneum Sampling to Assess Bioequivalence between Topical Acyclovir Products. Pharm. Res. 2019, 36, 180.
- Bielfeldt, S.; Bonnier, F.; Byrne, H.J.; Chourpa, I.; Dancik, Y.; Lane, M.E.; Lunter, D.J.; Munnier, E.; Puppels, G.; Tfayli, A.; et al. Monitoring dermal penetration and permeation kinetics of topical products; the role of Raman microspectroscopy. TrAC Trends Anal. Chem. 2022, 156, 116709.
- Liu, Y.; Lunter, D.J. Confocal Raman spectroscopy at different laser wavelengths in analyzing stratum corneum and skin penetration properties of mixed PEGylated emulsifier systems. Int. J. Pharm. 2022, 616, 121561.
- Lunter, D.; Daniels, R. Confocal Raman microscopic investigation of the effectiveness of penetration enhancers for procaine delivery to the skin. J. Biomed. Opt. 2014, 19, 126015.
- Iliopoulos, F.; Tang, C.F.; Li, Z.; Rahma, A.; Lane, M.E. Confocal Raman Spectroscopy for Assessing Bioequivalence of Topical Formulations. Pharmaceutics 2023, 15, 1075.
More
Information
Subjects:
Dermatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.5K
Revisions:
2 times
(View History)
Update Date:
07 Jul 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No