 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Matteo Ponzoni | -- | 3583 | 2023-07-04 15:22:40 | | | |
| 2 | Lindsay Dong | -58 word(s) | 3525 | 2023-07-05 03:12:42 | | |
Video Upload Options
Even with modern therapy, patients with heart failure only have a 50% five-year survival rate. To improve the development of new therapeutic strategies, preclinical models of disease are needed to properly emulate the human condition. Determining the most appropriate model represents the first key step for reliable and translatable experimental research. Rodent models of heart failure provide a strategic compromise between human in vivo similarity and the ability to perform a larger number of experiments and explore many therapeutic candidates.
1. Introduction
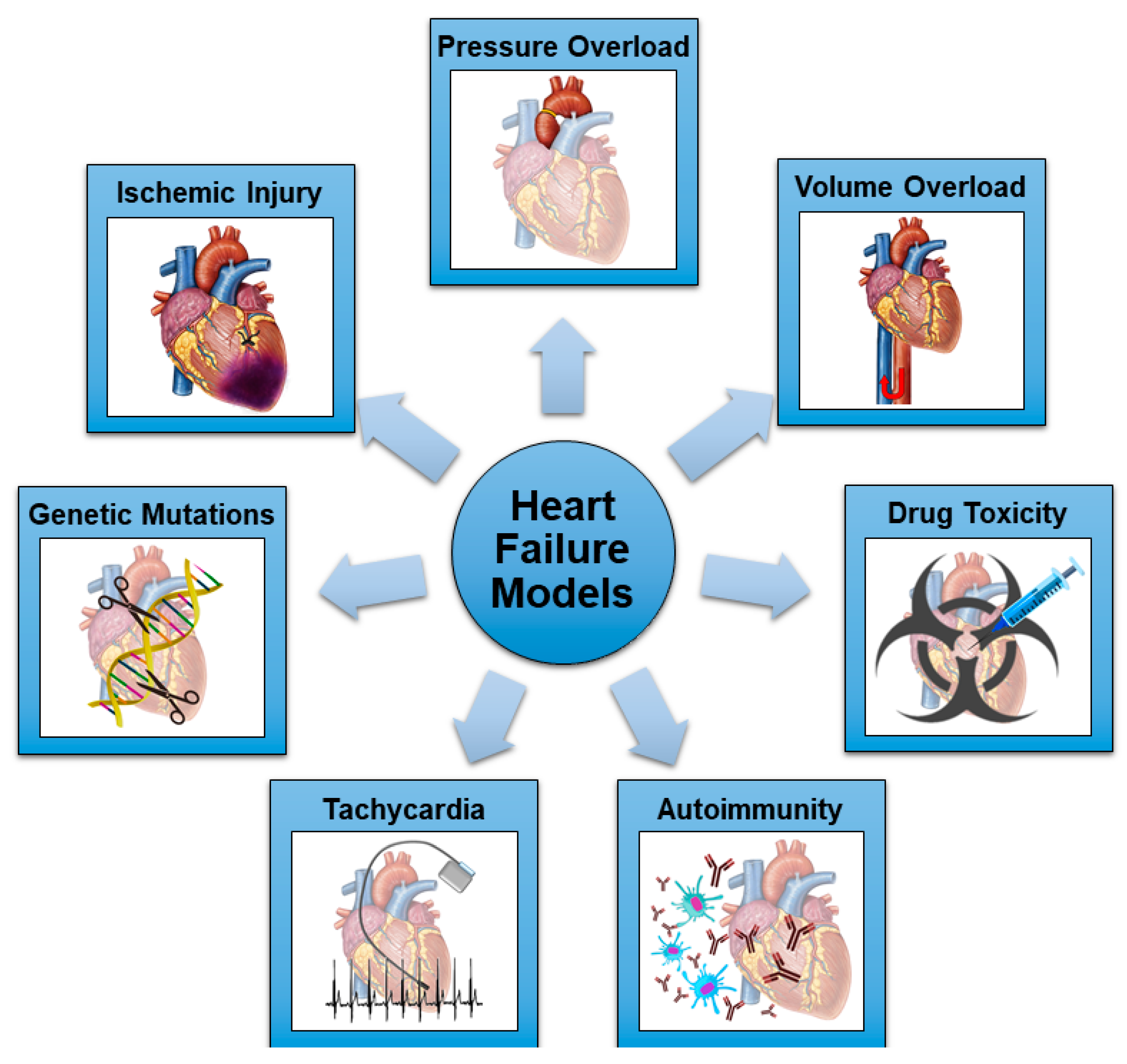
2. Rats and Mice as Animal Models of Heart Failure
3. General Considerations of Rat Models
4. Ischemic Injury Models
5. Pressure Overload Models
6. Volume Overload Models
7. Drug Toxicity Models
8. Autoimmune-Mediated Models
Acute autoimmune myocarditis has been reproduced in rats by injecting porcine myocardial myosin and generating a cross-reactivity with native cardiomyocytes. Using a two-stage protocol of subcutaneous footpad injection of purified porcine cardiac myosin supplemented with complete Freund adjuvant, cardiomyocyte injury, inflammatory infiltrate, and replacement fibrosis are achieved even 18 days after the completion of the injection protocol [76]. Extensive myocardial damage is sustained by several modalities of cell death (i.e., necroptosis, apoptosis, and autophagy) [77]. After 3 weeks, early signs of LV contractile impairment can be detected using cardiac magnetic resonance tissue tracking [78]. Four weeks after the completion of the protocol, LV dilatation and a drop in ejection fraction are evident with standard echocardiography [76]. Interestingly, the RV seems to be almost spared from foci of late gadolinium enhancement with magnetic resonance imaging, suggesting preferential effect on the LV from the acute myocarditis (inflammatory) process [79].
9. Rapid Ventricular Pacing Models
10. Genetic Models
11. Conclusions
References
- Truby, L.K.; Rogers, J.G. Advanced Heart Failure: Epidemiology, Diagnosis, and Therapeutic Approaches. JACC. Heart Fail. 2020, 8, 523–536.
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the Impact of Heart Failure in the United States: A Policy Statement from the American Heart Association. Circ. Heart Fail. 2013, 6, 606–619.
- Schilling, C.; Dalziel, K.; Nunn, R.; Du Plessis, K.; Shi, W.Y.; Celermajer, D.; Winlaw, D.; Weintraub, R.G.; Grigg, L.E.; Radford, D.J.; et al. The Fontan Epidemic: Population Projections from the Australia and New Zealand Fontan Registry. Int. J. Cardiol. 2016, 219, 14–19.
- Moons, P.; Bovijn, L.; Budts, W.; Belmans, A.; Gewillig, M. Temporal Trends in Survival to Adulthood among Patients Born with Congenital Heart Disease from 1970 to 1992 in Belgium. Circulation 2010, 122, 2264–2272.
- Rossano, J.W.; Cherikh, W.S.; Chambers, D.C.; Goldfarb, S.; Hayes, D.J.; Khush, K.K.; Kucheryavaya, A.Y.; Toll, A.E.; Levvey, B.J.; Meiser, B.; et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Twenty-First Pediatric Heart Transplantation Report-2018; Focus Theme: Multiorgan Transplantation. J. Hear. Lung Transplant. Off. Publ. Int. Soc. Hear. Transplant. 2018, 37, 1184–1195.
- Zangwill, S. Five Decades of Pediatric Heart Transplantation: Challenges Overcome, Challenges Remaining. Curr. Opin. Cardiol. 2017, 32, 69–77.
- Kirk, R.; Dipchand, A.I.; Davies, R.R.; Miera, O.; Chapman, G.; Conway, J.; Denfield, S.; Gossett, J.G.; Johnson, J.; McCulloch, M.; et al. ISHLT Consensus Statement on Donor Organ Acceptability and Management in Pediatric Heart Transplantation. J. Hear. lung Transplant. Off. Publ. Int. Soc. Hear. Transplant. 2020, 39, 331–341.
- Schlagenhauf, A.; Kalbhenn, J.; Geisen, U.; Beyersdorf, F.; Zieger, B. Acquired von Willebrand Syndrome and Platelet Function Defects during Extracorporeal Life Support (Mechanical Circulatory Support). Hamostaseologie 2020, 40, 221–225.
- Lukito, P.; Wong, A.; Jing, J.; Arthur, J.F.; Marasco, S.F.; Murphy, D.A.; Bergin, P.J.; Shaw, J.A.; Collecutt, M.; Andrews, R.K.; et al. Mechanical Circulatory Support Is Associated with Loss of Platelet Receptors Glycoprotein Ibα and Glycoprotein VI. J. Thromb. Haemost. 2016, 14, 2253–2260.
- Ishigami, S.; Ohtsuki, S.; Eitoku, T.; Ousaka, D.; Kondo, M.; Kurita, Y.; Hirai, K.; Fukushima, Y.; Baba, K.; Goto, T.; et al. Intracoronary Cardiac Progenitor Cells in Single Ventricle Physiology: The PERSEUS (Cardiac Progenitor Cell Infusion to Treat Univentricular Heart Disease) Randomized Phase 2 Trial. Circ. Res. 2017, 120, 1162–1173.
- Makkar, R.R.; Kereiakes, D.J.; Aguirre, F.; Kowalchuk, G.; Chakravarty, T.; Malliaras, K.; Francis, G.S.; Povsic, T.J.; Schatz, R.; Traverse, J.H.; et al. Intracoronary ALLogeneic Heart STem Cells to Achieve Myocardial Regeneration (ALLSTAR): A Randomized, Placebo-Controlled, Double-Blinded Trial. Eur. Heart J. 2020, 41, 3451–3458.
- Zhu, J.; Ide, H.; Fu, Y.Y.; Teichert, A.-M.; Kato, H.; Weisel, R.D.; Maynes, J.T.; Coles, J.G.; Caldarone, C.A. Losartan Ameliorates “Upstream” Pulmonary Vein Vasculopathy in a Piglet Model of Pulmonary Vein Stenosis. J. Thorac. Cardiovasc. Surg. 2014, 148, 2550–2557.
- Ponzoni, M.; Frigo, A.C.; Castaldi, B.; Cerutti, A.; Di Salvo, G.; Vida, V.L.; Padalino, M.A. Surgical Strategies for the Management of End-Stage Heart Failure in Infants and Children: A 15-Year Experience with a Patient-Tailored Approach. Artif. Organs 2021, 45, 1543–1553.
- Ponzoni, M.; Castaldi, B.; Padalino, M.A. Pulmonary Artery Banding for Dilated Cardiomyopathy in Children: Returning to the Bench from Bedside. Children 2022, 9, 1392.
- Traister, A.; Patel, R.; Huang, A.; Patel, S.; Plakhotnik, J.; Lee, J.E.; Medina, M.G.; Welsh, C.; Ruparel, P.; Zhang, L.; et al. Cardiac Regenerative Capacity Is Age- and Disease-Dependent in Childhood Heart Disease. PLoS ONE 2018, 13, e0200342.
- Zhang, L.; Yeganeh, A.; Bartkevics, M.; Perri, A.; Brown, C.; Coles, J.; Maynes, J.T. Mesenchymal Stromal Cells Isolated From Patients With Congenital Heart Disease Reveal an Age-Dependent Proinflammatory Phenotype. JACC Adv. 2022, 1, 100050.
- Wang, P.; Li, H.; Zhu, M.; Han, R.Y.; Guo, S.; Han, R. Correction of DMD in Human IPSC-Derived Cardiomyocytes by Base-Editing-Induced Exon Skipping. Mol. Ther.—Methods Clin. Dev. 2023, 28, 40–50.
- Nishiyama, T.; Zhang, Y.; Cui, M.; Li, H.; Sanchez-Ortiz, E.; McAnally, J.R.; Tan, W.; Kim, J.; Chen, K.; Xu, L.; et al. Precise Genomic Editing of Pathogenic Mutations in RBM20 Rescues Dilated Cardiomyopathy. Sci. Transl. Med. 2022, 14.
- Patten, R.D.; Hall-Porter, M.R. Small Animal Models of Heart Failure: Development of Novel Therapies, Past and Present. Circ. Heart Fail. 2009, 2, 138–144.
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal Models of Heart Failure: A Scientific Statement from the American Heart Association. Circ. Res. 2012, 111, 131–150.
- Riehle, C.; Bauersachs, J. Small Animal Models of Heart Failure. Cardiovasc. Res. 2019, 115, 1838–1849.
- Holtze, S.; Gorshkova, E.; Braude, S.; Cellerino, A.; Dammann, P.; Hildebrandt, T.B.; Hoeflich, A.; Hoffmann, S.; Koch, P.; Terzibasi Tozzini, E.; et al. Alternative Animal Models of Aging Research. Front. Mol. Biosci. 2021, 8, 660959.
- Makowski, M.R.; Wiethoff, A.J.; Jansen, C.H.P.; Botnar, R.M. Cardiovascular MRI in Small Animals. Expert Rev. Cardiovasc. Ther. 2010, 8, 35–47.
- Fu, X.; Segiser, A.; Carrel, T.P.; Tevaearai Stahel, H.T.; Most, H. Rat Heterotopic Heart Transplantation Model to Investigate Unloading-Induced Myocardial Remodeling. Front. Cardiovasc. Med. 2016, 3, 34.
- Dobson, G.P. On Being the Right Size: Heart Design, Mitochondrial Efficiency and Lifespan Potential. Clin. Exp. Pharmacol. Physiol. 2003, 30, 590–597.
- Ross, J.J. Dilated Cardiomyopathy: Concepts Derived from Gene Deficient and Transgenic Animal Models. Circ. J. 2002, 66, 219–224.
- Willott, R.H.; Gomes, A.V.; Chang, A.N.; Parvatiyar, M.S.; Pinto, J.R.; Potter, J.D. Mutations in Troponin That Cause HCM, DCM AND RCM: What Can We Learn about Thin Filament Function? J. Mol. Cell. Cardiol. 2010, 48, 882–892.
- Lee, Y.-K.; Jiang, Y.; Ran, X.-R.; Lau, Y.-M.; Ng, K.-M.; Lai, W.-H.K.; Siu, C.-W.; Tse, H.-F. Recent Advances in Animal and Human Pluripotent Stem Cell Modeling of Cardiac Laminopathy. Stem Cell Res. Ther. 2016, 7, 139.
- Yadav, S.; Sitbon, Y.H.; Kazmierczak, K.; Szczesna-Cordary, D. Hereditary Heart Disease: Pathophysiology, Clinical Presentation, and Animal Models of HCM, RCM, and DCM Associated with Mutations in Cardiac Myosin Light Chains. Pflug. Arch. 2019, 471, 683–699.
- Schultheiss, H.-P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated Cardiomyopathy. Nat. Rev. Dis. Prim. 2019, 5, 32.
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134.
- Faggiano, A.; Avallone, C.; Gentile, D.; Provenzale, G.; Toriello, F.; Merlo, M.; Sinagra, G.; Carugo, S. Echocardiographic Advances in Dilated Cardiomyopathy. J. Clin. Med. 2021, 10, 5518.
- 2014 ESC Guidelines on Diagnosis and Management of Hypertrophic Cardiomyopathy. Eur. Heart J. 2014, 35, 2733–2779.
- Rapezzi, C.; Aimo, A.; Barison, A.; Emdin, M.; Porcari, A.; Linhart, A.; Keren, A.; Merlo, M.; Sinagra, G. Restrictive Cardiomyopathy: Definition and Diagnosis. Eur. Heart J. 2022, 43, 4679–4693.
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS Expert Consensus Statement on Evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy. Hear. Rhythm 2019, 16, e301–e372.
- Pilati, M.; Rebonato, M.; Formigari, R.; Butera, G. Endomyocardial Biopsy in Pediatric Myocarditis and Dilated Cardiomyopathy: A Tool in Search for a Role. J. Cardiovasc. Dev. Dis. 2022, 9.
- RONA, G.; CHAPPEL, C.I.; BALAZS, T.; GAUDRY, R. An Infarct-like Myocardial Lesion and Other Toxic Manifestations Produced by Isoproterenol in the Rat. AMA. Arch. Pathol. 1959, 67, 443–455.
- Adler, N.; Camin, L.L.; Shulkin, P. Rat Model for Acute Myocardial Infarction: Application to Technetium-Labeled Glucoheptonate, Tetracycline, and Polyphosphate. J. Nucl. Med. 1976, 17, 203–207.
- Ryu, J.H.; Kim, I.-K.; Cho, S.-W.; Cho, M.-C.; Hwang, K.-K.; Piao, H.; Piao, S.; Lim, S.H.; Hong, Y.S.; Choi, C.Y.; et al. Implantation of Bone Marrow Mononuclear Cells Using Injectable Fibrin Matrix Enhances Neovascularization in Infarcted Myocardium. Biomaterials 2005, 26, 319–326.
- Pfeffer, M.A.; Pfeffer, J.M.; Fishbein, M.C.; Fletcher, P.J.; Spadaro, J.; Kloner, R.A.; Braunwald, E. Myocardial Infarct Size and Ventricular Function in Rats. Circ. Res. 1979, 44, 503–512.
- Kainuma, S.; Miyagawa, S.; Fukushima, S.; Tsuchimochi, H.; Sonobe, T.; Fujii, Y.; Pearson, J.T.; Saito, A.; Harada, A.; Toda, K.; et al. Influence of Coronary Architecture on the Variability in Myocardial Infarction Induced by Coronary Ligation in Rats. PLoS ONE 2017, 12, e0183323.
- Kolk, M.V.V.; Meyberg, D.; Deuse, T.; Tang-Quan, K.R.; Robbins, R.C.; Reichenspurner, H.; Schrepfer, S. LAD-Ligation: A Murine Model of Myocardial Infarction. J. Vis. Exp. 2009.
- Reichert, K.; Colantuono, B.; McCormack, I.; Rodrigues, F.; Pavlov, V.; Abid, M.R. Murine Left Anterior Descending (LAD) Coronary Artery Ligation: An Improved and Simplified Model for Myocardial Infarction. J. Vis. Exp. 2017.
- Yang, S.M.; Liu, J.; Li, C.X. Intermedin Protects against Myocardial Ischemia-Reperfusion Injury in Hyperlipidemia Rats. Genet. Mol. Res. 2014, 13, 8309–8319.
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of Neonatal and Adult Mammalian Heart Regeneration by the MiR-15 Family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192.
- Kalra, S.; Bhatt, H.; Kirtane, A.J. Stenting in Primary Percutaneous Coronary Intervention for Acute ST-Segment Elevation Myocardial Infarction. Methodist Debakey Cardiovasc. J. 2018, 14, 14–22.
- Vidal-Calés, P.; Cepas-Guillén, P.L.; Brugaletta, S.; Sabaté, M. New Interventional Therapies beyond Stenting to Treat ST-Segment Elevation Acute Myocardial Infarction. J. Cardiovasc. Dev. Dis. 2021, 8, 100.
- Michael, L.H.; Entman, M.L.; Hartley, C.J.; Youker, K.A.; Zhu, J.; Hall, S.R.; Hawkins, H.K.; Berens, K.; Ballantyne, C.M. Myocardial Ischemia and Reperfusion: A Murine Model. Am. J. Physiol. 1995, 269, H2147–H2154.
- Szabó, P.L.; Dostal, C.; Pilz, P.M.; Hamza, O.; Acar, E.; Watzinger, S.; Mathew, S.; Kager, G.; Hallström, S.; Podesser, B.K.; et al. Remote Ischemic Perconditioning Ameliorates Myocardial Ischemia and Reperfusion-Induced Coronary Endothelial Dysfunction and Aortic Stiffness in Rats. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 702–713.
- Rockman, H.A.; Wachhorst, S.P.; Mao, L.; Ross, J.J. ANG II Receptor Blockade Prevents Ventricular Hypertrophy and ANF Gene Expression with Pressure Overload in Mice. Am. J. Physiol. 1994, 266, H2468–H2475.
- Feldman, A.M.; Weinberg, E.O.; Ray, P.E.; Lorell, B.H. Selective Changes in Cardiac Gene Expression during Compensated Hypertrophy and the Transition to Cardiac Decompensation in Rats with Chronic Aortic Banding. Circ. Res. 1993, 73, 184–192.
- Miranda-Silva, D.; Gonçalves-Rodrigues, P.; Almeida-Coelho, J.; Hamdani, N.; Lima, T.; Conceição, G.; Sousa-Mendes, C.; Cláudia-Moura; González, A.; Díez, J.; et al. Characterization of Biventricular Alterations in Myocardial (Reverse) Remodelling in Aortic Banding-Induced Chronic Pressure Overload. Sci. Rep. 2019, 9, 2956.
- Gao, J.P.; Chen, C.X.; Wu, Q.; Gu, W.L.; Li, X. Effect of Sodium Houttuyfonate on Inhibiting Ventricular Remodeling Induced by Abdominal Aortic Banding in Rats. Can. J. Physiol. Pharmacol. 2010, 88, 693–701.
- Hirata, M.; Ousaka, D.; Arai, S.; Okuyama, M.; Tarui, S.; Kobayashi, J.; Kasahara, S.; Sano, S. Novel Model of Pulmonary Artery Banding Leading to Right Heart Failure in Rats. Biomed Res. Int. 2015, 2015, 753210.
- Nakao, Y.; Aono, J.; Hamaguchi, M.; Takahashi, K.; Sakaue, T.; Inoue, K.; Ikeda, S.; Yamaguchi, O. O-Ring-Induced Transverse Aortic Constriction (OTAC) Is a New Simple Method to Develop Cardiac Hypertrophy and Heart Failure in Mice. Sci. Rep. 2022, 12, 85.
- Andersen, S.; Schultz, J.G.; Holmboe, S.; Axelsen, J.B.; Hansen, M.S.; Lyhne, M.D.; Nielsen-Kudsk, J.E.; Andersen, A. A Pulmonary Trunk Banding Model of Pressure Overload Induced Right Ventricular Hypertrophy and Failure. J. Vis. Exp. 2018.
- Bossers, G.P.L.; Hagdorn, Q.A.J.; Koop, A.-M.C.; van der Feen, D.E.; Bartelds, B.; van Leusden, T.; De Boer, R.A.; Silljé, H.H.W.; Berger, R.M.F. Female Rats Are Less Prone to Clinical Heart Failure than Male Rats in a Juvenile Rat Model of Right Ventricular Pressure Load. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H994–H1002.
- Mendes-Ferreira, P.; Santos-Ribeiro, D.; Adão, R.; Maia-Rocha, C.; Mendes-Ferreira, M.; Sousa-Mendes, C.; Leite-Moreira, A.F.; Brás-Silva, C. Distinct Right Ventricle Remodeling in Response to Pressure Overload in the Rat. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H85–H95.
- Nishimura, K.; Oydanich, M.; Zhang, J.; Babici, D.; Fraidenraich, D.; Vatner, D.E.; Vatner, S.F. Rats Are Protected from the Stress of Chronic Pressure Overload Compared with Mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R894–R900.
- Litwin, S.E.; Katz, S.E.; Weinberg, E.O.; Lorell, B.H.; Aurigemma, G.P.; Douglas, P.S. Serial Echocardiographic-Doppler Assessment of Left Ventricular Geometry and Function in Rats with Pressure-Overload Hypertrophy. Chronic Angiotensin-Converting Enzyme Inhibition Attenuates the Transition to Heart Failure. Circulation 1995, 91, 2642–2654.
- Chen, J.; Chemaly, E.R.; Liang, L.F.; LaRocca, T.J.; Yaniz-Galende, E.; Hajjar, R.J. A New Model of Congestive Heart Failure in Rats. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H994–H1003.
- Ruppert, M.; Barta, B.A.; Korkmaz-Icöz, S.; Loganathan, S.; Oláh, A.; Sayour, A.A.; Benke, K.; Nagy, D.; Bálint, T.; Karck, M.; et al. Sex Similarities and Differences in the Reverse and Anti-Remodeling Effect of Pressure Unloading Therapy in a Rat Model of Aortic Banding and Debanding. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H204–H222.
- Miranda-Silva, D.G.; Rodrigues, P.; Alves, E.; Rizo, D.; Fonseca, A.C.R.G.; Lima, T.; Baganha, F.; Conceição, G.; Sousa, C.; Gonçalves, A.; et al. Mitochondrial Reversible Changes Determine Diastolic Function Adaptations During Myocardial (Reverse) Remodeling. Circ. Heart Fail. 2020, 13, e006170.
- Cho, J.S.; Lee, J.; Park, K.C.; Yang, K.-J.; Cho, E.J. The Relationship between MiRNA-26b and Connective Tissue Growth Factor in Rat Models of Aortic Banding and Debanding. Korean J. Intern. Med. 2021, 36, 596–607.
- Akazawa, Y.; Fujioka, T.; Ide, H.; Yazaki, K.; Honjo, O.; Sun, M.; Friedberg, M.K. Impaired Right and Left Ventricular Function and Relaxation Induced by Pulmonary Regurgitation Are Not Reversed by Tardive Antifibrosis Treatment. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H38–H51.
- Droogmans, S.; Franken, P.R.; Garbar, C.; Weytjens, C.; Cosyns, B.; Lahoutte, T.; Caveliers, V.; Pipeleers-Marichal, M.; Bossuyt, A.; Schoors, D.; et al. In Vivo Model of Drug-Induced Valvular Heart Disease in Rats: Pergolide-Induced Valvular Heart Disease Demonstrated with Echocardiography and Correlation with Pathology. Eur. Heart J. 2007, 28, 2156–2162.
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. Pharmacol. 2015, 70, 5.47.1–5.47.20.
- Andersen, A.; van der Feen, D.E.; Andersen, S.; Schultz, J.G.; Hansmann, G.; Bogaard, H.J. Animal Models of Right Heart Failure. Cardiovasc. Diagn. Ther. 2020, 10, 1561–1579.
- Traister, A.; Li, M.; Aafaqi, S.; Lu, M.; Arab, S.; Radisic, M.; Gross, G.; Guido, F.; Sherret, J.; Verma, S.; et al. Integrin-Linked Kinase Mediates Force Transduction in Cardiomyocytes by Modulating SERCA2a/PLN Function. Nat. Commun. 2014, 5, 4533.
- Khosroshahi, A.J.; Mokhtari, B.; Badalzadeh, R. Combination of Nicotinamide Mononucleotide and Troxerutin Induces Full Protection against Doxorubicin-Induced Cardiotoxicity by Modulating Mitochondrial Biogenesis and Inflammatory Response. Mol. Biol. Rep. 2022.
- O’Connell, J.L.; Romano, M.M.D.; Campos Pulici, E.C.; Carvalho, E.E.V.; de Souza, F.R.; Tanaka, D.M.; Maciel, B.C.; Salgado, H.C.; Fazan-Júnior, R.; Rossi, M.A.; et al. Short-Term and Long-Term Models of Doxorubicin-Induced Cardiomyopathy in Rats: A Comparison of Functional and Histopathological Changes. Exp. Toxicol. Pathol. Off. J. Ges. Fur Toxikol. Pathol. 2017, 69, 213–219.
- Chakouri, N.; Farah, C.; Matecki, S.; Amedro, P.; Vincenti, M.; Saumet, L.; Vergely, L.; Sirvent, N.; Lacampagne, A.; Cazorla, O. Screening for In-Vivo Regional Contractile Defaults to Predict the Delayed Doxorubicin Cardiotoxicity in Juvenile Rat. Theranostics 2020, 10, 8130–8142.
- Pandey, S.; Dvorakova, M.C. Future Perspective of Diabetic Animal Models. Endocr. Metab. Immune Disord.—Drug Targets 2020, 20, 25–38.
- Marchini, G.S.; Cestari, I.N.; Salemi, V.M.C.; Irigoyen, M.C.; Arnold, A.; Kakoi, A.; Rocon, C.; Aiello, V.D.; Cestari, I.A. Early Changes in Myocyte Contractility and Cardiac Function in Streptozotocin-Induced Type 1 Diabetes in Rats. PLoS ONE 2020, 15, e0237305.
- Lakomkin, V.L.; Abramov, A.A.; Lukoshkova, E.V.; Prosvirnin, A.V.; Kapelko, V.I. Systolic Dysfunction of the Heart in Type 1 Diabetes Mellitus. Bull. Exp. Biol. Med. 2021, 172, 14–17.
- Nana-Leventaki, E.; Nana, M.; Poulianitis, N.; Sampaziotis, D.; Perrea, D.; Sanoudou, D.; Rontogianni, D.; Malliaras, K. Cardiosphere-Derived Cells Attenuate Inflammation, Preserve Systolic Function, and Prevent Adverse Remodeling in Rat Hearts With Experimental Autoimmune Myocarditis. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 70–77.
- Wu, Y.; Zheng, Z.; Cao, X.; Yang, Q.; Norton, V.; Adini, A.; Maiti, A.K.; Adini, I.; Wu, H. RIP1/RIP3/MLKL Mediates Myocardial Function Through Necroptosis in Experimental Autoimmune Myocarditis. Front. Cardiovasc. Med. 2021, 8, 696362.
- Zhu, J.; Chen, Y.; Xu, Z.; Wang, S.; Wang, L.; Liu, X.; Gao, F. Non-Invasive Assessment of Early and Acute Myocarditis in a Rat Model Using Cardiac Magnetic Resonance Tissue Tracking Analysis of Myocardial Strain. Quant. Imaging Med. Surg. 2020, 10, 2157–2167.
- Korkusuz, H.; Esters, P.; Naguib, N.; Nour Eldin, N.-E.; Lindemayr, S.; Huebner, F.; Koujan, A.; Bug, R.; Ackermann, H.; Vogl, T.J. Acute Myocarditis in a Rat Model: Late Gadolinium Enhancement with Histopathological Correlation. Eur. Radiol. 2009, 19, 2672–2678.
- Farrag, N.A.; Thornhill, R.E.; Prato, F.S.; Skanes, A.C.; Sullivan, R.; Sebben, D.; Butler, J.; Sykes, J.; Wilk, B.; Ukwatta, E. Assessment of Left Atrial Fibrosis Progression in Canines Following Rapid Ventricular Pacing Using 3D Late Gadolinium Enhanced CMR Images. PLoS ONE 2022, 17, e0269592.
- Zhou, F.; Fu, W.-D.; Chen, L. MiRNA-182 Regulates the Cardiomyocyte Apoptosis in Heart Failure. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4917–4923.
- Sepúlveda, M.; Gonano, L.A.; Back, T.G.; Chen, S.R.W.; Vila Petroff, M. Role of CaMKII and ROS in Rapid Pacing-Induced Apoptosis. J. Mol. Cell. Cardiol. 2013, 63, 135–145.
- Aistrup, G.L.; Kelly, J.E.; Kapur, S.; Kowalczyk, M.; Sysman-Wolpin, I.; Kadish, A.H.; Wasserstrom, J.A. Pacing-Induced Heterogeneities in Intracellular Ca2+ Signaling, Cardiac Alternans, and Ventricular Arrhythmias in Intact Rat Heart. Circ. Res. 2006, 99, e65–e73.
- Greaser, M.L.; Warren, C.M.; Esbona, K.; Guo, W.; Duan, Y.; Parrish, A.M.; Krzesinski, P.R.; Norman, H.S.; Dunning, S.; Fitzsimons, D.P.; et al. Mutation That Dramatically Alters Rat Titin Isoform Expression and Cardiomyocyte Passive Tension. J. Mol. Cell. Cardiol. 2008, 44, 983–991.
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a Gene for Hereditary Cardiomyopathy, Regulates Titin Splicing. Nat. Med. 2012, 18, 766–773.
- Guo, W.; Zhu, C.; Yin, Z.; Zhang, Y.; Wang, C.; Walk, A.S.; Lin, Y.-H.; McKinsey, T.A.; Woulfe, K.C.; Ren, J.; et al. The Ryanodine Receptor Stabilizer S107 Ameliorates Contractility of Adult Rbm20 Knockout Rat Cardiomyocytes. Physiol. Rep. 2021, 9, e15011.
- Schiabor Barrett, K.M.; Cirulli, E.T.; Bolze, A.; Rowan, C.; Elhanan, G.; Grzymski, J.J.; Lee, W.; Washington, N.L. Cardiomyopathy Prevalence Exceeds 30% in Individuals with TTN Variants and Early Atrial Fibrillation. Genet. Med. 2023, 100012.
- Marcello, M.; Cetrangolo, V.; Savarese, M.; Udd, B. Use of Animal Models to Understand Titin Physiology and Pathology. J. Cell. Mol. Med. 2022, 26, 5103–5112.
- Steinhorn, B.; Sorrentino, A.; Badole, S.; Bogdanova, Y.; Belousov, V.; Michel, T. Chemogenetic Generation of Hydrogen Peroxide in the Heart Induces Severe Cardiac Dysfunction. Nat. Commun. 2018, 9, 4044.
- Sorrentino, A.; Steinhorn, B.; Troncone, L.; Saravi, S.S.S.; Badole, S.; Eroglu, E.; Kijewski, M.F.; Divakaran, S.; Di Carli, M.; Michel, T. Reversal of Heart Failure in a Chemogenetic Model of Persistent Cardiac Redox Stress. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H617–H626.
- Spyropoulos, F.; Sorrentino, A.; van der Reest, J.; Yang, P.; Waldeck-Weiermair, M.; Steinhorn, B.; Eroglu, E.; Saeedi Saravi, S.S.; Yu, P.; Haigis, M.; et al. Metabolomic and Transcriptomic Signatures of Chemogenetic Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H451–H465.

