Even with modern therapy, patients with heart failure only have a 50% five-year survival rate. To improve the development of new therapeutic strategies, preclinical models of disease are needed to properly emulate the human condition. Determining the most appropriate model represents the first key step for reliable and translatable experimental research. Rodent models of heart failure provide a strategic compromise between human in vivo similarity and the ability to perform a larger number of experiments and explore many therapeutic candidates. We herein review the currently available rodent models of heart failure, summarizing their physiopathological basis, the timeline of the development of ventricular failure, and their specific clinical features. In order to facilitate the future planning of investigations in the field of heart failure, a detailed overview of the advantages and possible drawbacks of each model is provided.
1. Introduction
Thanks to the advances in medical therapies for heart failure (HF), as well as the persistent improvements in cardiovascular surgical techniques and devices technologies, many previously fatal cardiac diseases are now chronically managed with satisfactory medium- and long-term prognosis. However, once the cardiac performance is severely affected, a progressively declining syndrome is established, whose definitive fate is often end-stage organ failure [
1]. Demographic projections estimate that by the next decade one out of every thirty-three people in the US will be affected by HF [
2]. In the highly complex population of pediatric patients with structural heart defects, the number of patients palliated with the Fontan procedure is expected to double in the next 20 years [
3]. Currently, more than 90% of children with congenital heart defects survive to adulthood [
4], indicating a substantial increase in demand for HF-related services as this population ages into adulthood with expected evolving cardiac dysfunction.
Current treatment options for end-stage HF entail organ replacement with human grafts, artificial devices, or xenografts. However, none these strategies are definitively curative (incorporating graft failure) and are limited by the availability of donors [
5], chronic immune rejection [
6,
7], high rates of graft vascular disease, and abnormal activation of coagulation cascades and platelets at the blood–device interface [
8,
9]. Conversely, regenerative medicine techniques may represent a new frontier in the management of end-stage HF. Cardiac progenitor stem cells and derived proteins [
10,
11], molecularly targeted drugs [
12], and reinvented surgical procedures [
13,
14] aim at stimulating the intrinsic repair ability of the human heart [
15,
16]. Genome editing technologies are emerging as potential therapeutic strategies able to address specific causative monogenetic disorders associated with the development of cardiomyopathy and HF. Through base editing, the expression of key proteins that are dysregulated in rare genetic forms of dilated cardiomyopathies (DCM), such as dystrophin in Duchenne muscular dystrophy [
17] and Rbm20 [
18], can be restored, representing a promising therapeutic concept.
To test novel putative therapeutic approaches to HF in a controlled manner, viable animal models of HF are paramount. Due to an advantageous compromise between adequate body size and low housing and maintenance costs, rodent models of HF have been extensively used by basic and translational scientists for decades [
19,
20,
21]. Particularly, rats offer sufficient dimensions to perform cardiac surgical procedures and invasive hemodynamic measurements safely, shortening learning curves for operators. Moreover, they expedite advanced imaging measurements and provide a 10-fold greater myocardial mass for subsequent histopathological or molecular analyses, when compared to mice.
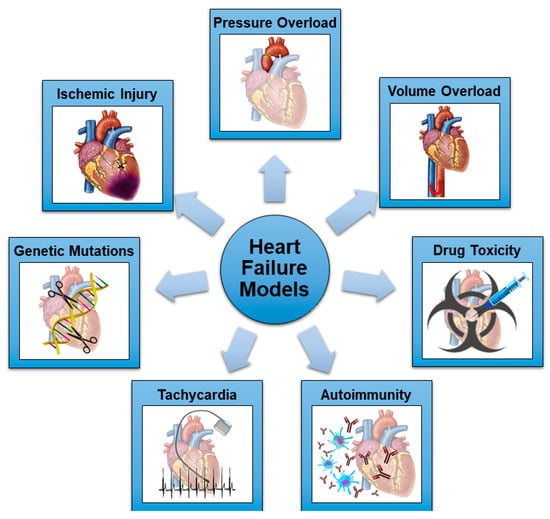
Many methods have been tested and validated to induce progressive HF and DCM in rats, mimicking the different etiologies of HF in humans. The schematic representation of the most common mechanisms utilized in rodent models in HF is shown in Figure 1.
Figure 1. Schematic representation of the most common mechanisms utilized in rodent models of HF.
2. Rats and Mice as Animal Models of Heart Failure
Rats and mice share the benefits of small mammalian models, in terms of ease of handling and housing, short breeding cycle, and the number of recruitable animals to improve the statistical power of experiments. The small physical size reduces the costs of novel therapeutic agents and molecules, whose administration is usually calculated based on body weight. However, some differences apply between these two species, starting with the size of the animals. Despite a similar 2–3 years life span, adult (6–8 week-old) rats weigh 250 to 550 g, while adult mice are generally 10 times smaller (25–30 g) [
22]. This aspect becomes particularly valuable in the setting of experimental surgery. Animal intubation and ventilation, access to anatomical structures of interest, and performing specific procedures are easier in larger rodents, shortening learning times for operators and increasing animal survival. Moreover, invasive micromanometer catheterization can be accomplished more safely in rats, and the spectrum of applicable noninvasive echocardiographic and magnetic resonance imaging techniques mimics the one available in the clinical setting [
23,
24]. Finally, a greater number of different postmortem analyses can be accomplished on the same rat, thanks to the larger size of tissues and higher intravascular blood volume.
Rodent cardiac physiology, contraction patterns, and energetics resemble the human equivalents. Rodent and human myocardial tissue share similar functions for many proteins, although rodent cardiomyocytes exhibit a predominance of alpha-myosin heavy chains (compared to beta-myosin heavy chains in humans), which are characterized by a rapid ATPase activity to facilitate the extremely high heart rate and the short cardiac cycle. Mice present a resting heart rate of 500 to 600 bpm, with 350 bpm for rats. Although still far from human values, the de-escalation from mice to rats in terms of cellular bioenergetics and mitochondrial efficiency contributes to translational value [
25]. These differences should be taken into account particularly in the setting of preventative and reparative molecular therapies for HF since they could influence the likelihood of moving findings into human clinical practice [
21]. An intermediate validation in large animal models is mandatory for this purpose (i.e., porcine).
The high degree of genetic similarity between rodents and humans allowed for the creation of relevant transgenic and knockout strains with an HF phenotype [
21]. Easier manipulation of the mouse genome explains the extremely large number of transgenic mouse strains currently available for target analysis. Mouse models obtained from mutations in cardiac myosin light chain, lamin, troponin, and other extra or intrasarcomeric proteins have been addressed in [
28,
29,
30,
31]. Conversely, genetic rat models of HF are infrequent [
21]. The translation of molecular mediators resulting from genetically modulated rodents into human studies requires a cautious approach. Genetic mutations that cause idiopathic DCM in humans are mostly undiscovered [
32]. Although many proteins share similar functions, their expression levels and the final organ and biological effects can differ substantially between rodents and larger mammals and humans [
19,
21].
3. General Considerations of Rat Models
The ideal animal model should be able to reproduce the typical echocardiographic, histological, and clinical features of the desired type of HF. Reflecting the higher prevalence of left-sided HF in the general population, left ventricular (LV) dysfunction models leading to HF and eventually DCM are more widely utilized. Human DCM is defined as a spectrum of myocardial diseases which share ventricular dilatation and depressed contractility [
34]. The key phenotype is characterized by a progressive LV dilatation, together with a ventricular shape transition from its original ellipsoid shape to a more spherical one, wall thinning, and a global reduction in contractility, which is revealed by a decrease in stroke volume, cardiac index, and increased strain parameters [
32,
35]. These features differentiate DCM from other cardiomyopathies, such as hypertrophic cardiomyopathy (where increased LV wall thickness and normal or even supranormal contractility is noted [
36]), restrictive cardiomyopathy (in which ventricular chamber dimensions are reduced, impairing LV filling and creating a primary diastolic dysfunction [
37]), and arrhythmogenic right ventricular cardiomyopathy (characterized by typical electrocardiographic anomalies and an often pathognomonic fibrous-fatty myocardial replacement [
38]).
In DCM, associated diastolic dysfunction can occur, and the combination of increased LV filling pressures and ventricular dilatation often generates functional mitral regurgitation. Magnetic resonance imaging can detect areas of late gadolinium enhancement, which represent the process of diffuse fibrosis that is common to many DCM etiologies (the slower heart rate of the rat also makes cardiac MRI more feasible, although specialized instrumentation is still needed). Endomyocardial biopsy typically reveals morphological alterations in DCM: myocardial disarray, fibrosis, cell death, cardiomyocyte hypertrophy, scar formation, and inflammatory infiltration [
45]. Importantly, this phenotype could characterize only the LV or present with biventricular involvement, depending on the etiology and severity of the disease. This aspect requires particular attention when choosing the most adequate animal model for the desired experimentation.
To date, a benchmark rat model of DCM is not available. Existing animal models are not able to respond to all of the above-mentioned requisites of the ideal model. The main reason lies in the fundamental difference in the development of human DCM versus the induction of experimental DCM. As previously stated, DCM in humans has a wide spectrum of causes, but in most of the cases a leading factor cannot be detected, often with evolution over significant time periods. Experimental DCM models rely mostly on a single pathogenic pathway with relatively rapid onset, which can consequently reproduce only some specific features of the DCM phenotype.
4. Ischemic Injury Models
Ischemic injury in rats has been induced in different ways: subcutaneous injections of isoproterenol [
49], direct damage using electrocautery [
50], arterial ligation, or cryogenic damage with a metal probe cooled in liquid nitrogen [
51]. In 1979, Pfeffer et al. introduced the left anterior descending (LAD) artery ligation model [
55], which now has become the preferred ischemic model. A direct correlation exists between the infarct size and the severity of LV dilatation and contractile function impairment, which ranges from completely preserved if the scar involves <30% of the LV circumference to congestive HF if >46% [
55]. However, the predictability of the infarcted myocardial area after LAD ligation is less consistent and varies depending on the level where the suture is placed and anatomical variation [
56]. Coronary anatomy in rats presents significant variability between animals: the single septal branch may originate from the proximal left coronary artery in 60% of cases and from the proximal right coronary in the remaining 40%; the circumflex artery branches distally from a long left main coronary artery in 66% of animals, while it arises more proximally from a short left main coronary artery or the main septal branch in 34% [
56]. As a result, a distal LAD ligation always creates an infarction only in the LV anterior wall, while a proximal ligation (just below the left atrial appendage) can create a wider anterolateral infarction (64% of cases) or an only anterior infarction (36%). Standardized surgical protocols are now widely available to guide the investigator [
53,
54], helping to reduce model animal-to-animal variability.
An alternative ischemic model consists of ligation of the circumflex artery, which is demonstrated to produce a significant infarct zone (40% of LV diameter) [
52]. However, this procedure is less validated and, being technically more demanding, should be adopted as a secondary option to the LAD ligation model.
A general concern regarding the ischemic rat model of HF is that the LAD ligation procedure is usually performed in young (4–8-week-old) rats, while ischemic diseases affect an older and multidiseased population. Since the regenerative potential of the human and mammalian heart is age dependent [
15,
134], and the causes of idiopathic DCM rely only marginally on an ischemic substrate, the transition from the LAD ligation model to clinical practice necessitates careful verifications in this specific context. On the other hand, the leading cause of congestive HF in humans remains coronary arterial disease, which the LAD ligation model clearly exemplifies. Thanks to the advancement of emergency care, most of the patients that present with an acute myocardial infarction with ST-elevation can now benefit from a prompt revascularization strategy [
135]. Despite successful revascularization, up to 20% of patients surviving an ST-elevation myocardial infarction are hospitalized with HF in the first year after the event [
136]. After revascularization, the underlying pathophysiological basis of myocardial damage switches from irreversible ischemia and cell necrosis to transient ischemia and ischemia-reperfusion injury. To account for these mechanisms, the LAD ligation procedure has evolved from a permanent ligation to a temporary (typically 30 min) ligation, followed by controlled reperfusion [
57]. This approach allowed for the investigation of molecular pathways involved in the protective role of the ischemic preconditioning process [
58].
5. Pressure Overload Models
Pressure overload can be generated in rats using surgical or non-surgical methods (Table 1). Surgical techniques usually entail an abrupt augmentation of the ventricular afterload by placing a tight band around the aorta or the pulmonary artery (PA).
Aortic banding can be achieved at different levels. In the original procedure described by Rockman et al. in a mouse model [
60], a suture is placed around the transverse aortic arch and tightened against a 27 G needle. The same technique has been implemented in rats (using an 18–22 G needle) and modified by applying a more proximal banding of the ascending aorta [
62,
63], which mimics the pathophysiological features of LV failure induced by severe aortic stenosis. Finally, the abdominal aorta can be banded to investigate the mechanisms of ventricular remodeling by creating a slower-developing hypertensive HF profile [
68].
Tightening of the aorta or PA can be accomplished using a suture tied against a needle, with the needle rapidly removed to restore antegrade blood flow. Given the need for complete vessel mobilization and the related risk of fatal bleedings, alternative techniques have been proposed, such as half-closed surgical clips [
69], which generate even higher pressure gradients across the PA with minimal mobilization of the vessel, or O-rings of predetermined diameters, which guarantee reproducible grades of aortic banding in mice [
61].
Using these surgical approaches, delineated RV failure can be achieved 7 weeks from PA banding, as demonstrated by a reduced ejection fraction and cardiac output and severely dilated right chambers [
70], with temporal sex-related differences in developing the HF phenotype (observed less contractile and diastolic dysfunction and fibrosis in females) [
71]. The grade of PA banding contributes directly to the timing of the transition to a decompensated RV response, which can start manifesting after 1–3 weeks from the procedure in case of severe PA constriction [
70,
72]. On the other hand, aortic banding in rats produces an initial compensated hypertrophic response, with preserved ejection fraction and LV diameters 4 weeks after banding [
64], which can last until 18–20 weeks before LV systo-diastolic failure occurs [
62,
65,
66]. Interestingly, this adaptation to pressure overload is generally absent in mice, which develop very early LV failure and DCM [
64]. Understanding the timing of progression from hypertrophy to cardiac decompensation in rat models is mandatory to guide the interventional and analytical planning of experiments.
Although technically challenging and with not insignificant mortality rates [
66], rats can undergo aortic de-banding to investigate the antiremodeling effects of LV unloading which are accomplished by aortic valve replacement or ventricular assist devices in the clinical setting. After 6 to 9 weeks of aortic banding, LV unloading (i.e., de-banding) promotes the regression of hypertrophy and the recovery of diastolic function [
73], supported by the restoration of mitochondrial energetics [
74] and a reduction of pro-fibrotic factors [
67]. Interestingly, aortic banding triggers significant systo-diastolic impairment and activation of prohypertrophic and profibrotic pathways also in the RV, which persist partially altered even after de-banding [
63].
6. Volume Overload Models
Only one rat model of pulmonary regurgitation is currently available in the scientific literature. By lacerating the pulmonary leaflets with a 22.5 G needle inserted into the main PA through a purse-string suture, pulmonary regurgitation was achieved by Akazawa et al. [
99]. In this model, RV dilatation manifested after only 2 weeks, while progressive RV systolic dysfunction occurred after 4 weeks. Due to negative ventricular–ventricular interactions, RV dilatation contributes to diastolic LV compression and impaired relaxation. Interestingly, cardiomyocyte hypertrophy and myocardial fibrosis affected the RV exclusively [
99], underlying differential biological pathways supporting ventricular–ventricular interactions in response to RV volume and pressure overload.
Finally, multivalvular regurgitation involving all four cardiac valves has been produced by long-term pergolide and serotonin administration in rats [
100], simulating carcinoid heart disease in advanced neuroendocrine tumors.
7. Drug Toxicity Models
Drug administration can induce HF in experimental rat models by direct toxicity of myocardial tissue (doxorubicin, ethanol, and homocysteine), producing myocardial ischemia (isoproterenol), type 1 diabetes mellitus (streptozotocin [
139]), or pulmonary hypertension (monocrotaline [
79]).
Repeated intraperitoneal doxorubicin injections represent one of the most validated rodent models of HF progressing to DCM [
140]. By generating mitochondrial dysfunction and intense oxidative stress [
101], doxorubicin promotes swelling and vacuolization of cardiomyocytes, disorganization of myofibrils, and intense interstitial fibrosis [
102]. Different administration protocols have been described, although a long-term scheme with intraperitoneal weekly injections for a total of 9 weeks (cumulative dose of 18mg/kg) has been found to be more effective to create LV dilatation and systolic impairment [
102]. Dose-dependent cardiotoxicity is present, together with a regionalized effect on myocardial contraction, whose earliest impairment is noticed in the basal LV segments by speckle tracking imaging [
103].
Several rodent models of diabetes mellitus are available [
143]. These models have contributed to the discovery and validation of many antidiabetic molecules and the investigation of diabetes-related multi-organ complications. The intraperitoneal injection of streptozotocin causes a reproducible chemical ablation of pancreatic beta cells, inducing irreversible diabetes in rats. Early signs of diabetic cardiomyopathy can be documented 2–3 weeks after the injection, such as prolonged contraction and relaxation times in isolated cardiomyocytes and enlarged left atrium and impaired systolic parameters with echocardiography [
112,
113].
8. Autoimmune-Mediated Models
Acute autoimmune myocarditis has been reproduced in rats by injecting porcine myocardial myosin and generating a cross-reactivity with native cardiomyocytes. Using a two-stage protocol of subcutaneous footpad injection of purified porcine cardiac myosin supplemented with complete Freund adjuvant, cardiomyocyte injury, inflammatory infiltrate, and replacement fibrosis are achieved even 18 days after the completion of the injection protocol [117]. Extensive myocardial damage is sustained by several modalities of cell death (i.e., necroptosis, apoptosis, and autophagy) [118]. After 3 weeks, early signs of LV contractile impairment can be detected using cardiac magnetic resonance tissue tracking [119]. Four weeks after the completion of the protocol, LV dilatation and a drop in ejection fraction are evident with standard echocardiography [117]. Interestingly, the RV seems to be almost spared from foci of late gadolinium enhancement with magnetic resonance imaging, suggesting preferential effect on the LV from the acute myocarditis (inflammatory) process [120].
9. Rapid Ventricular Pacing Models
Due to practical reasons, rapid pacing-induced HF models usually involve larger mammals, in whom a pacemaker with a pacing lead in the RV apex is implanted subcutaneously and used to maintain supranormal heart rates for several weeks [
150]. Rapid pacing has also been adopted in rats to investigate the intracellular effects of chronic tachycardia. Zhou et al. surgically implanted an electrode on the epicardial surface of the RV apex of rats and used it for rapid pacing (550 bpm) for 4 weeks with an external pacemaker [
122]. Tachycardia and rapid pacing promoted rat myocyte apoptotic pathways, increased the intracellular levels of reactive oxygen species [
121], and dysregulated calcium signaling, especially at higher pacing rates [
123]. These cellular mechanisms translated into the development of an apoptotic-based HF profile, with reduced LV contractile function and increased LV end-diastolic pressure after 4 weeks of rapid pacing [
122]. Given the need for extracorporeal devices, rapid pacing rat models might be more suitable for the in vitro study of single myocardial fiber response to chronic electric stimulation than whole animal in vivo experiments.
10. Genetic Models
Although murine models of HF induced by specific genetic mutations are widely available, few genetically based rat models have been reported. Greaser et al. identified a rat strain with an autosomal dominant mutation of the gene encoding the RNA binding motif protein 20 (Rbm20), which alters the isoform expression of the sarcomeric protein titin (TTN) [
124]. Titin is a cardiac and skeletal muscle protein involved in sarcomere assembly and protection from overstretching. Both heterozygous and homozygous Rbm20-deficient rats exhibit a phenotype of DCM with dilated LV, increased subendocardial fibrosis, but initially preserved contractile function [
125]. Starting from 3 months of age, progressive LV wall thinning and a decrease in fractional shortening and cardiac output are observed [
126]. As seen in humans with hereditary DCM caused by Rbm20 mutations, fibrosis is accompanied by electrical abnormalities that predispose to arrhythmias and sudden cardiac death, which start at 10 months of age in rats [
125].
Given its central role in controlling the mechanical properties of the sarcomere, truncating variants of TTN lead to a wide number of inherited myopathies and cardiac disorders [
151], ranging from hypertrophic to dilated ventricular profiles. Several mouse models carrying variants in the TTN gene have been developed to understand the structural role of TTN and the clinical correlates in patients. All of the available animal models have been excellently summarized by Marcello and colleagues in a very recent review exclusively focused on TTN pathophysiology [
152].
Recently, a novel rat model that relies on chemo–genetic interactions was established by Steinhorn et al., injecting rats with adeno-associated virus type 9 carrying a cardiac-specific recombinant D-amino acid oxidase, which produces hydrogen peroxidase during the conversion of D-amino acids into alpha-keto acids [
131]. When rats are fed with D-alanine (the substrate of the enzyme D-amino acid oxidase), the acute generation of intracellular hydrogen peroxidase induces a state of oxidative stress in cardiomyocytes. After 4 weeks of the specific diet, rats present HF with reduced LV contractile function and an enlarged LV. Interestingly, LV thickness is maintained and histopathological analysis confirms the absence of fibrosis (at 4 weeks, fibrosis develops later at 8 weeks) [
131,
133]. Cardiac transcriptome and metabolome analysis revealed marked alterations in mitochondrial function, cardiac energetics, redox homeostasis, amino acid metabolism, cytoskeletal and extracellular matrix organization, and antioxidant systems pathways [
132]. This HF model was noticeably reverted by the administration of an angiotensin II receptor blocker, which normalized the echocardiographic, morphologic, and metabolomic markers of disease [
132]. Oxidative stress is a common finding in different etiologies of HF, and this model may facilitate the testing of new molecular targets potentially involved in the process of ventricular remodeling.
11. Conclusions
Adopting the most representative animal model provides the basis for reliable experimental research that translates to clinical practice. Rats provide a strategic compromise between sufficient body size for several morphological evaluations/ease of interventions and low maintenance costs/short breeding cycles. Understanding the advantages and possible drawbacks of each specific rat model can expedite experiments, improve reproducibility, and strengthen reliability.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24043162