Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Emanuele Bernardinelli | -- | 1213 | 2023-07-04 11:23:07 | | | |
| 2 | Sirius Huang | Meta information modification | 1213 | 2023-07-05 02:47:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bernardinelli, E.; Huber, F.; Roesch, S.; Dossena, S. POU3F4-Linked Hearing Loss. Encyclopedia. Available online: https://encyclopedia.pub/entry/46386 (accessed on 27 July 2026).
Bernardinelli E, Huber F, Roesch S, Dossena S. POU3F4-Linked Hearing Loss. Encyclopedia. Available at: https://encyclopedia.pub/entry/46386. Accessed July 27, 2026.
Bernardinelli, Emanuele, Florian Huber, Sebastian Roesch, Silvia Dossena. "POU3F4-Linked Hearing Loss" Encyclopedia, https://encyclopedia.pub/entry/46386 (accessed July 27, 2026).
Bernardinelli, E., Huber, F., Roesch, S., & Dossena, S. (2023, July 04). POU3F4-Linked Hearing Loss. In Encyclopedia. https://encyclopedia.pub/entry/46386
Bernardinelli, Emanuele, et al. "POU3F4-Linked Hearing Loss." Encyclopedia. Web. 04 July, 2023.
Copy Citation
X-linked deafness (DFNX) is estimated to account for up to 2% of cases of hereditary hearing loss and occurs in both syndromic and non-syndromic forms. POU3F4 is the gene most commonly associated with X-linked deafness (DFNX2, DFN3) and accounts for about 50% of the cases of X-linked non-syndromic hearing loss.
POU3F4
X-linked deafness
transcription factor
hearing loss
gene variants
1. Introduction
Hearing loss is the most common sensory defect and affects up to 5 in 1000 newborns worldwide in a disabling form [1]. The aetiology of hearing loss is multifactorial and includes genetic and environmental factors. Deafness can be the result of physical trauma, infections, or as a side effect of some medications such as diuretics, anti-cancer drugs, and aminoglycoside antibiotics, but it is estimated that genetic causes are responsible for at least 60% of the cases [2]. Multiple genes have been associated with hereditary hearing loss in a syndromic or non-syndromic form. Defects in genes found on the X chromosome are causative of X-linked hearing loss, which accounts for up to 2% of the cases of hereditary deafness and is characterized by a typical inheritance pattern. POU3F4 is the gene most commonly associated with X-linked hearing loss and accounts for about 50% of these cases [3].
2. X-Linked Hearing Loss
Most of the genes and loci associated with hearing loss are found on the 22 autosomal chromosomes, but a small percentage of causative genes can also be found on the sex chromosomes. X-linked deafness (DFNX) is estimated to account for up to 2% of the cases of hereditary deafness [3][4]. Due to the pattern of inheritance, hemizygous male individuals are most commonly affected by X-linked deafness, while female heterozygous carriers might display varying degrees of hearing loss. Both syndromic and non-syndromic forms of hearing loss are associated with X-chromosome alterations. Up to date, 17 X-chromosome genes have been associated with a syndromic form of hearing loss and, including COL4A5, which causes Alport syndrome, which is characterized by glomerulopathy, hearing loss, and lens abnormalities [5], and AIFM1, which is responsible for Charcot-Marie-Tooth syndrome, which is characterized by heterogeneous forms of motor and sensory neuropathies [6]. Some genes, such as COL4A6, PRSP1, and AIFM1, have been identified as causative of both syndromic and non-syndromic X-linked hearing loss [3]. For what concerns X-linked non-syndromic hearing loss, 5 genes have been identified so far: PRPS1 [7], SMPX [8], AIFM1 [9], COL4A6 [10], and POU3F4 [11]. The latter is the gene most commonly responsible for X-linked hearing loss and accounts for about 50% of the cases of X-linked non-syndromic hearing loss [3].
3. The Transcription Factor POU3F4
The gene coding for the transcription factor POU3F4/BRN4 (OMIM *300039) was identified in 1995 [12] as the gene responsible for X-linked mixed deafness with stapes fixation (DFNX2, DFN3, OMIM #304400). Later, the clinical features of POU3F4-related hearing loss have been better defined by linking defects in the transcription factor with specific inner and middle ear malformations, including an incomplete partition (IP) of the cochlea and thickening of the stapedial footplate, occasionally associated with an enlargement of the vestibular aqueduct (EVA) [13]. Another feature that is often associated with defects in POU3F4 is the occurrence of a cerebrospinal fluid gusher during inner ear surgery [14][15], which will be detailed below.
The transcription factor POU3F4 is expressed in the mesenchymal cells of the neural tube during early embryonic development in mice. It contributes to the embryogenesis and development of the otic capsule into the mature inner and middle ear, including the morphogenesis of the stapedial ossicle [16] and the fasciculation of the spiral ganglion neurons to eventually form the cochlear synapses and the surrounding sensory hair cells [17]. In the mouse model, POU3F4 also promotes the assembly and organization of connexins into plaques, which establish a tight connection between the supporting cells in the cochlea and are essential for an efficient circulation of K+ ions and other small metabolites [18]. K+ concentration in the endolymph is a crucial factor for the conversion of the mechanical stimulus elicited by sound into the electrical impulse that travels from the hearing organ to the brain. The lack of POU3F4 expression in the mouse model has been shown to result in the degeneration of connexin plaques and defects in the composition of the endolymphatic fluid, with loss of the endocochlear potential [18].
4. Clinical and Anatomical Features of POU3F4-Linked Hearing Loss
As it is a form of X-linked hearing loss, males are more often affected by POU3F4-linked hearing loss. Hemizygous males carrying a defective POU3F4 form typically show a severe degree of sensorineural or mixed hearing loss, while most heterozygous females do not display any significant hearing defect. Nonetheless, a number of cases of female heterozygous carriers displaying hearing loss and vestibular dysfunctions have also been reported [19][20].
The type of hearing loss reported in most cases is bilateral, severe to profound, with conductive and sensorineural components. The source of the conductive component is attributed to the defect and fixation of the stapes [21]. ENT surgeons consistently advise against stapedotomy due to the high frequency of severe cerebrospinal fluid (CSF) gusher and resulting loss of cochlear function. Cochlear implant is possible in POU3F4 patients; however, specific complications and adverse events such as CSF gusher and misplacement of the electrode into the internal auditory canal (IAC) need to be anticipated and possibly managed intraoperatively [14][21].
The typical morphology of the inner ear in POU3F4 patients has been described as incomplete partition type III (IP-III), first reported by Phelps et al. in 1991 [22] and later classified more concisely by Sennaroglu and Bajin in 2017 [23]. Due to an impaired embryogenic development of the otic capsule in comparison to a healthy cochlea (Figure 1A,B), the modiolus and the base of the cochlea are mostly missing but the interscalar septa are still present and the dimensions of the cochlea are roughly normal (Figure 1C,D). A missing anatomic separation of the cochlea from the IAC is also a typical hallmark of POU3F4-related hearing loss, and it can explain the potential risk of CSF gusher following iatrogenic opening of the cochlea. The vestibular aqueduct is described to show variable degrees of dilatation [21]; however, definite EVA is not found consistently in POU3F4/IP-III patients [24][25]. Still, an EVA has been reported in association with POU3F4 variants in a number of cases [14][25][26]; therefore, with the present knowledge, it is difficult to establish whether POU3F4 variants are causative of EVA or a contributing factor. The cochlear nerve can be found intact in all patients, making a cochlear implant supply for POU3F4 patients, albeit challenging, always possible.
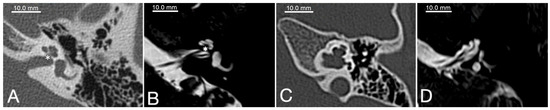
Figure 1. Radiological appearance of a normal cochlea (A,B) in comparison to a case of incomplete partition type III (C,D). All images represent axial planes of a left temporal bone. (A) and (C) are CT-scans; (B) and (D) are MRI T2-weighed axial scans. The cochlear base (*) can be clearly identified in (A) and (B) (normal cochlea), whereas it is missing in (C) and (D) (IP-III cochlea).
Besides hearing loss, further clinical symptoms are described in individual cases, such as peripheral vestibular dysfunction [27] as well as cognitive and developmental issues, ranging from attention disorders to delayed development, dystaxia, or hemiparalysis [14]. These findings gave rise to a discussion on whether DFNX2 should always be considered non-syndromic hearing loss. However, unfortunately, these symptoms are not consistently reported or systematically investigated, and the information remains sparse.
References
- Koffler, T.; Ushakov, K.; Avraham, K.B. Genetics of Hearing Loss: Syndromic. Otolaryngol. Clin. N. Am. 2015, 48, 1041–1061.
- Pandya, A. Genetic hearing loss: The journey of discovery to destination—How close are we to therapy? Mol. Genet. Genom. Med. 2016, 4, 583–587.
- Corvino, V.; Apisa, P.; Malesci, R.; Laria, C.; Auletta, G.; Franze, A. X-Linked Sensorineural Hearing Loss: A Literature Review. Curr. Genom. 2018, 19, 327–338.
- Del Castillo, I.; Morín, M.; Domínguez-Ruiz, M.; Moreno-Pelayo, M.A. Genetic etiology of non-syndromic hearing loss in Europe. Hum. Genet. 2022, 141, 683–696.
- Barker, D.F.; Hostikka, S.L.; Zhou, J.; Chow, L.T.; Oliphant, A.R.; Gerken, S.C.; Gregory, M.C.; Skolnick, M.H.; Atkin, C.L.; Tryggvason, K. Identification of Mutations in the COL4A5 Collagen Gene in Alport Syndrome. Science 1990, 248, 1224–1227.
- Poretti, A.; Palla, A.; Tarnutzer, A.A.; Petersen, J.A.; Weber, K.P.; Straumann, D.; Jung, H.H. Vestibular impairment in patients with Charcot-Marie-Tooth disease. Neurology 2013, 80, 2099–2105.
- Liu, X.; Han, D.; Li, J.; Han, B.; Ouyang, X.; Cheng, J.; Li, X.; Jin, Z.; Wang, Y.; Bitner-Glindzicz, M.; et al. Loss-of-Function Mutations in the PRPS1 Gene Cause a Type of Nonsyndromic X-linked Sensorineural Deafness, DFN2. Am. J. Hum. Genet. 2010, 86, 65–71.
- Huebner, A.K.; Gandia, M.; Frommolt, P.; Maak, A.; Wicklein, E.M.; Thiele, H.; Altmüller, J.; Wagner, F.; Viñuela, A.; Aguirre, L.A.; et al. Nonsense Mutations in SMPX, Encoding a Protein Responsive to Physical Force, Result in X-Chromosomal Hearing Loss. Am. J. Hum. Genet. 2011, 88, 621–627.
- Wang, Q.J.; Li, Q.Z.; Rao, S.Q.; Lee, K.; Huang, X.S.; Yang, W.Y.; Zhai, S.Q.; Guo, W.W.; Guo, Y.F.; Yu, N.; et al. AUNX1, a novel locus responsible for X linked recessive auditory and peripheral neuropathy, maps to Xq23-27.3. J. Med. Genet. 2006, 43, e33.
- Rost, S.; Bach, E.; Neuner, C.; Nanda, I.; Dysek, S.; Bittner, R.E.; Keller, A.; Bartsch, O.; Mlynski, R.; Haaf, T.; et al. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur. J. Hum. Genet. 2014, 22, 208–215.
- Cremers, C.W.; Snik, A.; Huygen, P.; Joosten, F.; Cremers, F. X-Linked Mixed Deafness Syndrome with Congenital Fixation of the Stapedial Footplate and Perilymphatic Gusher (DFN3). Adv. Otorhinolaryngol. 2002, 61, 161–167.
- De Kok, Y.J.; van der Maarel, S.M.; Bitner-Glindzicz, M.; Huber, I.; Monaco, A.P.; Malcolm, S.; Pembrey, M.E.; Ropers, H.H.; Cremers, F.P. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 1995, 267, 685–688.
- Gong, W.-X.; Gong, R.-Z.; Zhao, B. HRCT and MRI findings in X-linked non-syndromic deafness patients with a POU3F4 mutation. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1756–1762.
- Stankovic, K.M.; Hennessey, A.M.; Herrmann, B.; Mankarious, L.A. Cochlear Implantation in Children with Congenital X-Linked Deafness Due to Novel Mutations in POU3F4 Gene. Ann. Otol. Rhinol. Laryngol. 2010, 119, 815–822.
- Sennaroğlu, L.; Bajin, M.D. Incomplete partition type III: A rare and difficult cochlear implant surgical indication. Auris Nasus Larynx 2018, 45, 26–32.
- Samadi, D.S.; Saunders, J.C.; Crenshaw, E.B., 3rd. Mutation of the POU-domain gene Brn4/Pou3f4 affects middle-ear sound conduction in the mouse. Hear. Res. 2005, 199, 11–21.
- Brooks, P.M.; Rose, K.P.; MacRae, M.; Rangoussis, K.M.; Gurjar, M.; Hertzano, R.; Coate, T.M. Pou3f4-expressing otic mesenchyme cells promote spiral ganglion neuron survival in the postnatal mouse cochlea. J. Comp. Neurol. 2020, 528, 1967–1985.
- Kidokoro, Y.; Karasawa, K.; Minowa, O.; Sugitani, Y.; Noda, T.; Ikeda, K.; Kamiya, K. Deficiency of Transcription Factor Brn4 Disrupts Cochlear Gap Junction Plaques in a Model of DFN3 Non-Syndromic Deafness. PLoS ONE 2014, 9, e108216.
- Barashkov, N.A.; Klarov, L.A.; Teryutin, F.M.; Solovyev, A.V.; Pshennikova, V.G.; Konnikova, E.E.; Romanov, G.P.; Tobokhov, A.V.; Morozov, I.V.; Bondar, A.A.; et al. A novel pathogenic variant c.975G>A (p.Trp325*) in the POU3F4 gene in Yakut family (Eastern Siberia, Russia) with the X-linked deafness-2 (DFNX2). Int. J. Pediatr. Otorhinolaryngol. 2018, 104, 94–97.
- Marlin, S.; Moizard, M.; David, A.; Chaissang, N.; Raynaud, M.; Jonard, L.; Feldmann, D.; Loundon, N.; Denoyelle, F.; Toutain, A. Phenotype and genotype in females with POU3F4 mutations. Clin. Genet. 2009, 76, 558–563.
- Sennaroglu, L.; Ozbal Batuk, M.; Bajin, M.D. Incomplete Partition Type III. In Inner Ear Malformations: Classification, Evaluation and Treatment; Sennaroglu, L., Ed.; Springer: International Publishing: Cham, Switzerland, 2022; pp. 271–282.
- Phelps, P.D.; Reardon, W.; Pembrey, M.; Bellman, S.; Luxom, L. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology 1991, 33, 326–330.
- Sennaroğlu, L.; Bajin, M.D. Classification and Current Management of Inner Ear Malformations. Balk. Med. J. 2017, 34, 397–411.
- Roesch, S.; Rasp, G.; Sarikas, A.; Dossena, S. Genetic Determinants of Non-Syndromic Enlarged Vestibular Aqueduct: A Review. Audiol. Res. 2021, 11, 423–442.
- Bernardinelli, E.; Roesch, S.; Simoni, E.; Marino, A.; Rasp, G.; Astolfi, L.; Sarikas, A.; Dossena, S. Novel POU3F4 variants identified in patients with inner ear malformations exhibit aberrant cellular distribution and lack of SLC6A20 transcriptional upregulation. Front. Mol. Neurosci. 2022, 15, 999833.
- Huang, B.-Q.; Zeng, J.-L.; Yuan, Y.-Y.; Dai, P. A novel mutation in POU3F4 in a Chinese family with X-linked non-syndromic hearing loss. J. Otol. 2015, 10, 78–82.
- Wang, A.; Shearer, A.E.; Zhou, G.W.; Kenna, M.; Poe, D.; Licameli, G.R.; Brodsky, J.R. Peripheral Vestibular Dysfunction Is a Common Occurrence in Children with Non-syndromic and Syndromic Genetic Hearing Loss. Front. Neurol. 2021, 12, 714543.
More
Information
Subjects:
Otorhinolaryngology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
636
Revisions:
2 times
(View History)
Update Date:
05 Jul 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No