 +1 credit
+1 credit
Video Upload Options
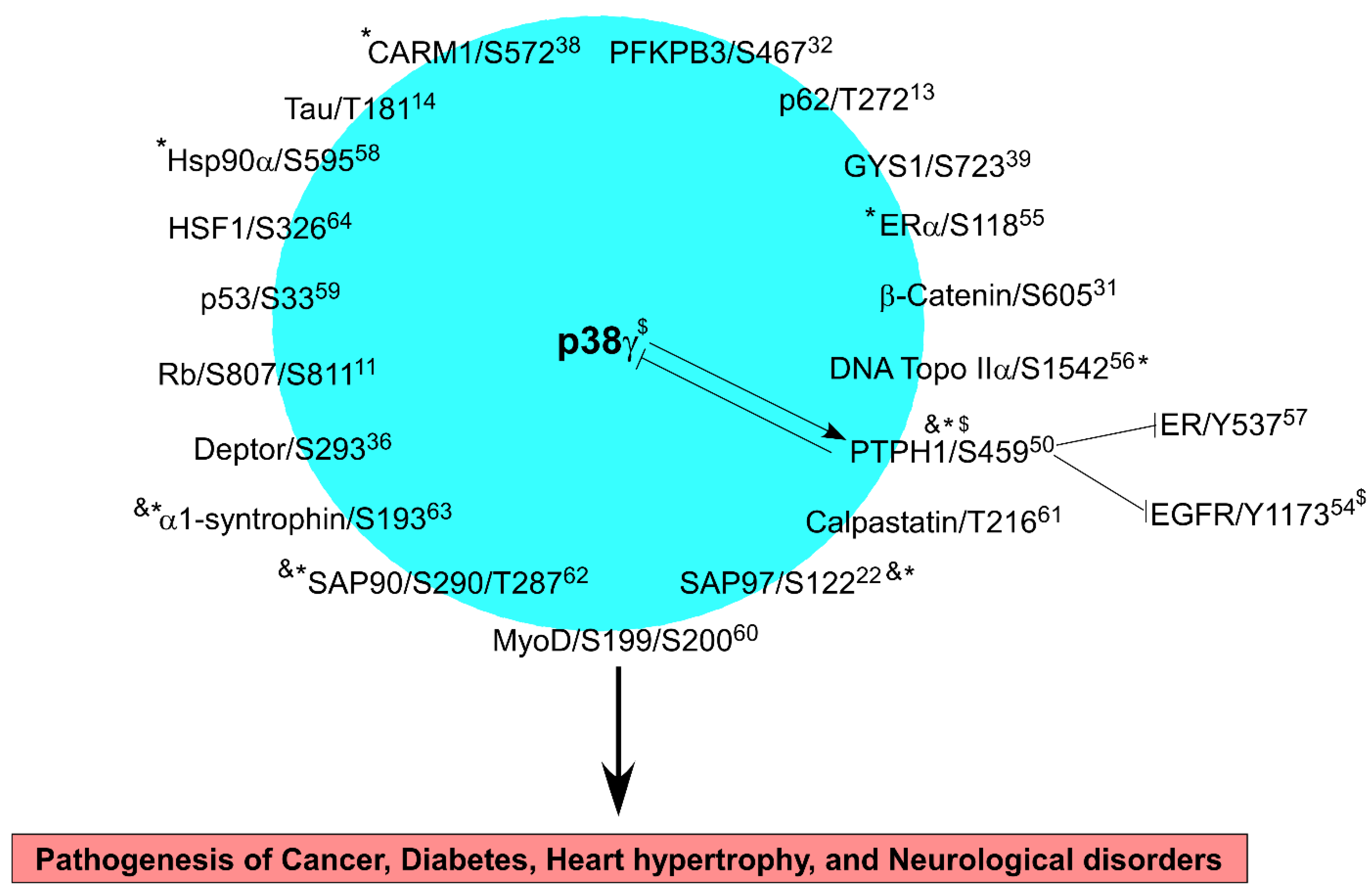
p38γ MAPK (also called ERK6 or SAPK3) is a family member of stress-activated MAPKs and has common and specific roles as compared to other p38 proteins in signal transduction. In addition to inflammation, p38γ metabolic signaling is involved in physiological exercise and in pathogenesis of cancer, diabetes, and Alzheimer’s disease, indicating its potential as a therapeutic target. p38γphosphorylates at least 19 substrates through which p38γ activity is further modified to regulate life-important cellular processes such as proliferation, differentiation, cell death, and transformation, thereby impacting biological outcomes of p38γ-driven pathogenesis. P38γ signaling is characterized by its unique reciprocal regulation with its specific phosphatase PTPH1 and by its direct binding to promoter DNAs, leading to transcriptional activation of targets including cancer-like stem cell drivers.
1. Introduction
2. p38γ Signaling to Its Substrates in Physiology and Diseases
References
- Ono, K.; Han, J. The p38 signal transduction pathway. Activation and function. Cell. Sign. 2000, 12, 1–13.
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 1–21.
- Qi, X.M.; Wang, F.; Chen, G. p38 Gamma MAPK Encyclopedia of Signaling Molecules, 2nd ed.; Choi, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 3718–3727.
- Cuenda, A.; Sanz-Ezquerro, J. p38g and p38d: From spectators to key physiological players. Trends Biochem. Sci. 2017, 42, 431–442.
- Pramanik, R.; Qi, X.; Borowicz, S.; Choubey, D.; Schultz, R.M.; Han, J.; Chen, G. p38 isoforms have opposite effects on AP-1-dependent transcription through regulation of c-Jun: The determinant role of the isoforms in the p38 MAPK signal specificity. J. Biol. Chem. 2003, 278, 4831–4839.
- Loesch, M.; Chen, G. The p38 MAPK stress pathway as a tumor suppressor or more? Front. Biosci. 2008, 13, 3581–3593.
- Qin, J.; Xin, H.; Qi, X.; Chen, G. Isoform-specific and cell/tissue-dependent effects of p38 MAPKs in regulating inflammation and inflammation-associated oncogenesis. Front. Biosci. Landmark 2022, 27, 031.
- Li, Z.; Jiang, Y.; Ulevitch, R.J.; Han, J. The primary structure of p38g: A new member of p38 group of MAP kinases. Biochem. Biophys. Res. Commun. 1996, 228, 334–340.
- Lechner, C.; Zahalka, M.A.; Giot, J.; Moller, N.P.; Ullrich, A. ERK6, a mitogen-activated protein kinase involved in C2C12 myoblast differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 4355–4359.
- Mertens, S.; Craxton, M.; Goedert, M. SAP kinase-3, a new member of the family of mammalian stress-activated protein kinases. FEBS Lett. 1996, 383, 273–276.
- Tomas-Loba, A.; Manieri, E.; Gonzalez-Teran, B.; Mora, A.; Leiva-Vega, L.; Santamans, A.M. p38g is essential for cell cycle progression and liver tumorigenesis. Nature 2019, 568, 557–560.
- Tang, J.; Qi, X.; Mercola, D.; Han, J.; Chen, G. Essential role of p38g in K-Ras transformation independent of phosphorylation. J. Biol. Chem. 2005, 280, 23910–23917.
- Pohl, N.M.; Qi, X.; Loesch, M.; Tang, J.; Li, Q.; Chen, G. Tissue-specific roles of p38g MAPK in Ras transformation. AACR Proc. 2006, 47, 2534.
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549.
- Hou, S.W.; Zhi, H.Y.; Pohl, N.; Loesch, M.; Qi, X.M.; Li, R.S. PTPH1 Dephosphorylates and Cooperates with p38γ MAPK to Increase Ras Oncogenesis through PDZ-Mediated Interaction. Cancer Res. 2010, 70, 2910.
- Sabio, G.; Simon, J.; Arthur, C.; Kuma, Y.; Peggie, M.; Carr, J.; Murray-Tait, V.; Centeno, F.; Goebeler, M.; Morrice, N.; et al. p38g regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 2005, 24, 1134–1145.
- Xu, M.; Ren, Z.; Wang, X.; Comer, A.; Frank, J.A.; Ke, Z. ErB2 and p38g MAPK mediate alcohol-induced increase in breast cancer stem cells. Mol. Cancer 2016, 15, 52.
- Hou, S.; Lepp, A.; Chen, G. p38 gamma MAP kinase. UCSD-Nat. Mol. Pages 2010.
- Hou, S.W.; Suresh, P.S.; Qi, X.; Lepp, A.; Mirza, S.; Chen, G. p38g Mitogen-activated Protein Kinase Signals through Phosphorylating Its Phosphatase PTPH1 in Regulating Ras Protein Oncogenesis and Stress Response. J. Biol. Chem. 2012, 287, 27895–27905.
- Zhi, H.; Hou, S.W.; Li, R.; Basir, Z.; Xiang, A.; Szabo, A. PTPH1 cooperates with vitamin D receptor to stimulate breast cancer growth through their mutual stabilization. Oncogene 2011, 30, 1706–1715.
- Ma, S.; Yin, N.; Qi, X.; Pfister, S.L.; Zhang, M.; Ma, R.; Chen, G. Tyrosine dephosphorylation enhances the therapeutic target activity of epidermal growth factor receptor (EGFR) by disrupting its interaction with estrogen receptor (ER). Oncotarget 2015, 6, 13320–13333.
- Chen, K.; Lin, S.; Wu, M.; Ho, M.; Santhanam, A.; Chou, C.; Meng, T.; Wang, A. Reciprocal allosteric regulation of p38g and PTPN3 involves a PDZ domain-modulated complex formation. Sci. Signal. 2014, 7, ra98.
- Yin, N.; Lepp, A.; Ji, Y.; Mortensen, M.; Hou, S.; Qi, X. The K-Ras effector p38g MAPK confers intrinsic resistance to tyrosine kinase inhibitors by stimulating EGFR transcription and EGFR dephosphorylation. J. Biol. Chem. 2017, 292, 15070–15079.
- Qi, X.; Wang, F.; Mortensen, M.; Wertz, R.; Chen, G. Targeting an oncogenic kinase/phosphatase signaling network for cancer therapy. Acta Pharm. Sin. B 2018, 8, 511–517.
- Qi, X.; Zhi, H.; Lepp, A.; Wang, P.; Huang, J.; Basir, Z. p38g mitogen-activated protein kinase (MAPK) confers breast cancer hormone sensitivity by switching estrogen receptor (ER) signaling from classical to nonclassical pathway via stimulating ER phosphorylation and c-Jun transcription. J. Biol. Chem. 2012, 287, 14681–14691.
- Qi, X.; Hou, S.; Lepp, A.; Li, R.; Basir, Z.; Lou, Z.; Chen, G. Phosphorylation and stabilization of topoisomerase IIa protein by p38g mitogen-activated protein kinase sensitize breast cancer cells to its poisons. J. Biol. Chem. 2011, 286, 35883–35890.
- Suresh, P.S.; Ma, S.; Migliaccio, A.; Chen, G. Protein-tyrosine phosphatase H1 increases breast cancer sensitivity to antiestrogens by dephosphorylating estrogen receptor at tyr537. Mol. Cancer Ther. 2014, 13, 230–238.
- Qi, X.M.; Xie, C.; Hou, S.; Li, G.; Yin, N.; Dong, L.; Lepp, A.; Chesnik, M.A.; Mirza, S.P.; Szabo, A.; et al. Identification of a ternary protein-complex as a therapeutic target for K-Ras-dependent colon cancer. Oncotarget 2014, 5, 4269–4282.
- Yin, N.; Qi, X.; Tsai, S.; Lu, Y.; Basir, Z.; Oshima, K. p38g MAPK is required for inflammation-associated colon tumorigenesis. Oncogene 2016, 35, 1039–1048.
- Wang, F.; Qi, X.; Wertz, R.; Mortensen, M.; Hagen, C.; Evans, J. p38g MAPK is essential for aerobic glycolysis and pancreatic tumorigenesis. Cancer Res. 2020, 80, 3251–3264.
- Kwong, J.; Hong, L.; Liao, R.; Deng, Q.; Han, J.; Sun, P. p38a and p38g mediates oncogenic ras-induced senescence through different mechanisms. J. Biol. Chem. 2009, 284, 11237–11246.
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A. Site-specific phosphorylation of tau inhibits amyloid-b toxicity in alzheimer’s mice. Science 2016, 354, 904–908.
- Chang, N.; Sincennes, M.; Chevalier, F.P.; Brun, C.E.; Lacaria, M.; Segales, J. The dystrophin glycoprotein complex regulates the epigenetic activation of muscle stem cell commitment. Cell Metab. 2019, 22, 755–768.
- Gonzalez-Teran, B.; Lopez, J.A.; Rodriguez, E.; Leiva, L.; Martinez-Martinez, S.; Bernal, J.A. p38g and d promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation. Nat. Commun. 2016, 7, 10477.
- Gillespie, M.A.; Grand, F.L.; Scime, A.; Kuang, S.; von Maltzahn, J.; Seale, V.; Cuenda, A.; Ranish, J.A.; Rudnicki, M.A. p38g-dependent gene silencing restricts entry into the myogenic differentiation program. J. Cell Biol. 2009, 187, 991–1005.
- Koh, A.; Molinaro, A.; Stahlman, M.; Khan, M.T.; Schmidt, C.; Manneras-Holm, L. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell 2018, 175, 947–961.
- Santamans, A.M.; Montalvo-Romeral, V.; Mora, A.; Lopez, J.A.; Gonzalez-Romero, F.; Jimenez-Blasco, D. p38γ and p38δ regulate postnatal cardiac metabolism through glycogen synthase 1. PLoS Biol. 2021, 19, e3001447.
- Loonat, A.A.; Martin, E.D.; Sarafraz-Shekary, N.; Tilgner, K.; Hertz, N.T.; Levin, R. p38g MAPK contributes to left ventricular remodeling after pathologic stress and disinhibits calpain through phosphorylation of calpastatin. FASEB J. 2019, 33, 13131–13144.
- Sabio, G.; Reuver, S.; Feijoo, C.; Hasegawa, M.; Thomas, G.M.; Centeno, F.; Kuhlendahl, S.; Leal-Ortiz, S.; Goedert, M.; Garner, C.; et al. Stress- and mitogen-induced phosphorylation of the synapse-associated protein SAP90/PSD-95 by activation of SAPK3/p38g and ERK1/ERK2. Biochem. J. 2004, 380, 19–30.
- Hasegawa, M.; Cuenda, A.; Spillantini, M.G.; Thomas, G.M.; Buee-Scherrer, V.; Cohen, P.; Goedert, M. Stress-activated protein kinase-3 interacts with the PDZ domain of a1-syntrophin: A mechanism for specific substrate recognition. J. Biol. Chem. 1999, 274, 12626–12631.
- Naidu, S.D.; Sutherland, C.; Zhang, Y.; Risco, A.; Vega, L.D.L.; Caunt, C.J. Heat shock factor 1 is a substrate for p38 mitogen-activated protein kinases. Mol. Cell. Biol. 2016, 36, 2403–2417.


