 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Nikolaos-Andreas Anastasopoulos | -- | 3397 | 2023-07-02 12:39:34 | | | |
| 2 | Peter Tang | + 1 word(s) | 3398 | 2023-07-03 05:18:19 | | |
Video Upload Options
Hepatocellular carcinoma (HCC) represents a worryingly increasing cause of malignancy-related mortality, while Metabolic Associated Fatty Liver Disease (MAFLD) is going to become its most common cause. Understanding the complex underlying pathophysiology of MAFLD-related HCC can provide opportunities for successful targeted therapies. Of particular interest in this sequela of hepatopathology is cellular senescence, a complex process characterised by cellular cycle arrest initiated by a variety of endogenous and exogenous cell stressors. A key biological process in establishing and maintaining senescence is oxidative stress, which is present in multiple cellular compartments of steatotic hepatocytes. Oxidative stress-induced cellular senescence can change hepatocyte function and metabolism, and alter, in a paracrine manner, the hepatic microenvironment, enabling disease progression from simple steatosis to inflammation and fibrosis, as well as HCC. The duration of senescence and the cell types it affects can tilt the scale from a tumour-protective self-restricting phenotype to the creator of an oncogenic hepatic milieu.
1. Oxidative Stress and Liver Metabolic Disease
1.1. The Oxygen Paradox
1.2. ROS in Various Biological Roles
1.3. ROS Regulation and Removal—Oxidative Eustress and Distress
1.4. The Role of Iron in OS
1.5. OS in Liver Metabolic Disease—The Concept of Lipotoxicity
2. Cellular Senescence
2.1. Overview of CS
2.2. CS and Molecular Players
3. MAFLD Specific Considerations
3.1. CS in MAFLD
3.2. CS and OS in MAFLD
3.3. Mitochondrial Dysfunction as a Link between OS and CS in MAFLD
4. Does CS Protect from or Induce Hepatocarcinogenesis in the Context of MAFLD?
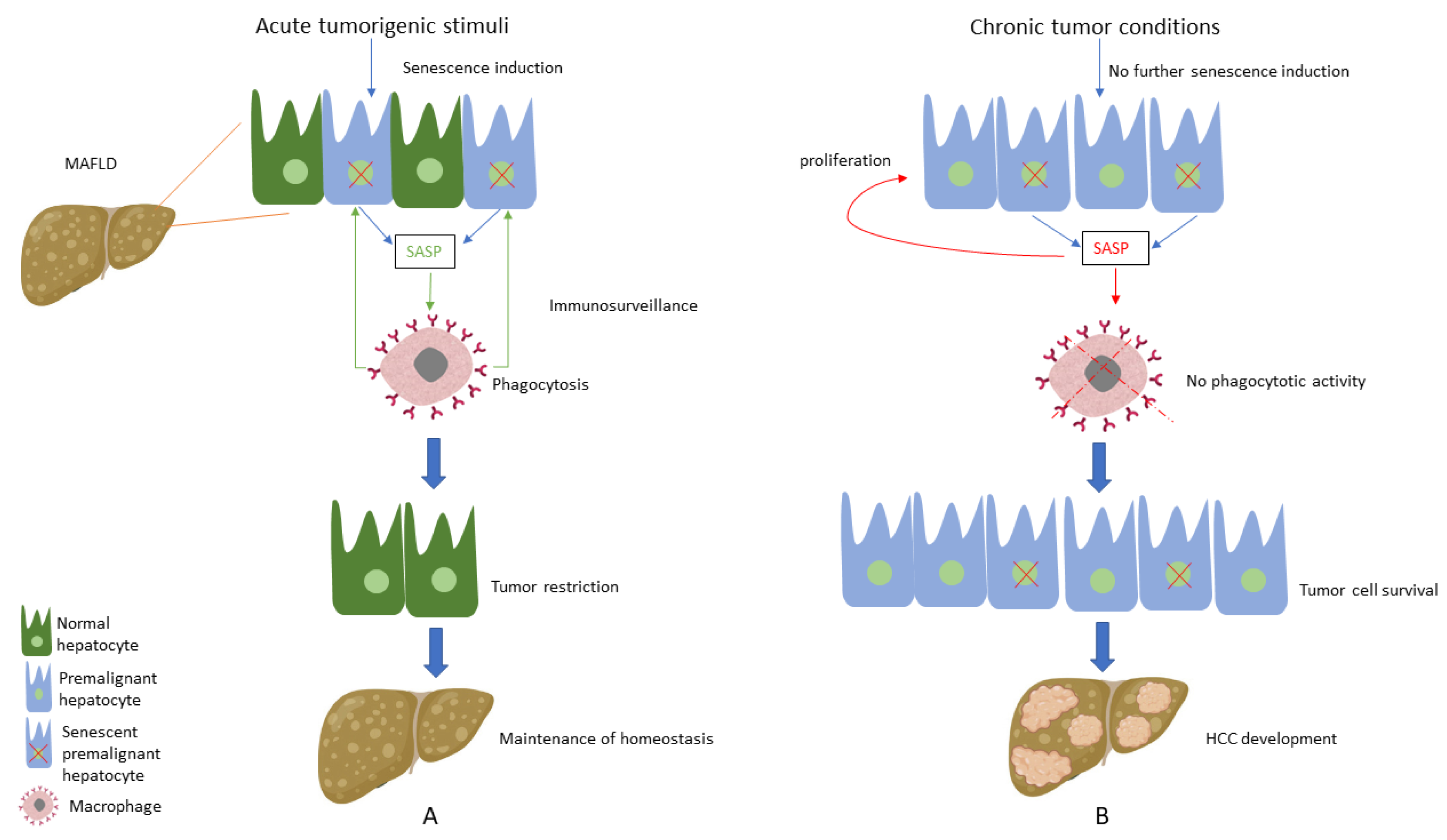
References
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron Homeostasis and Oxidative Stress: An Intimate Relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535.
- Barbouti, A.; Lagopati, N.; Veroutis, D.; Goulas, V.; Evangelou, K.; Kanavaros, P.; Gorgoulis, V.G.; Galaris, D. Implication of Dietary Iron-Chelating Bioactive Compounds in Molecular Mechanisms of Oxidative Stress-Induced Cell Ageing. Antioxidants 2021, 10, 491.
- Davies, K.J. Oxidative Stress: The Paradox of Aerobic Life. Biochem. Soc. Symp. 1995, 61, 1–31.
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Arauz, J.; Ramos-Tovar, E.; Muriel, P. Redox State and Methods to Evaluate Oxidative Stress in Liver Damage: From Bench to Bedside. Ann. Hepatol. 2016, 15, 160–173.
- Varesi, A.; Chirumbolo, S.; Campagnoli, L.I.M.; Pierella, E.; Piccini, G.B.; Carrara, A.; Ricevuti, G.; Scassellati, C.; Bonvicini, C.; Pascale, A. The Role of Antioxidants in the Interplay between Oxidative Stress and Senescence. Antioxidants 2022, 11, 1224.
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300.
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 716–748.
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147.
- Chandrasekaran, A.; Sosa, P.; Melendez, J.A. Redox Control of Senescence and Age-Related Disease. Redox Biol. 2017, 11, 91–102.
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic Syndrome, Aging and Involvement of Oxidative Stress. Aging Dis. 2015, 6, 109–120.
- Yan, F.; Li, K.; Xing, W.; Dong, M.; Yi, M.; Zhang, H. Role of Iron-Related Oxidative Stress and Mitochondrial Dysfunction in Cardiovascular Diseases. Oxid. Med. Cell Longev. 2022, 2022, 5124553.
- Mantzaris, M.D.; Bellou, S.; Skiada, V.; Kitsati, N.; Fotsis, T.; Galaris, D. Intracellular Labile Iron Determines H2O2-Induced Apoptotic Signaling via Sustained Activation of ASK1/JNK-P38 Axis. Free Radic. Biol. Med. 2016, 97, 454–465.
- Kitsati, N.; Mantzaris, M.D.; Galaris, D. Hydroxytyrosol Inhibits Hydrogen Peroxide-Induced Apoptotic Signaling via Labile Iron Chelation. Redox Biol. 2016, 10, 233–242.
- Anastasopoulos, N.-A.T.; Lianos, G.D.; Tatsi, V.; Karampa, A.; Goussia, A.; Glantzounis, G.K. Clinical Heterogeneity in Patients with Non-Alcoholic Fatty Liver Disease-Associated Hepatocellular Carcinoma. Expert. Rev. Gastroenterol. Hepatol. 2020, 14, 1025–1033.
- Lipke, K.; Kubis-Kubiak, A.; Piwowar, A. Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge. Cells 2022, 11, 844.
- Salekeen, R.; Haider, A.N.; Akhter, F.; Billah, M.M.; Islam, M.E.; Didarul Islam, K.M. Lipid Oxidation in Pathophysiology of Atherosclerosis: Current Understanding and Therapeutic Strategies. Int. J. Cardiol. Cardiovasc. Risk Prev. 2022, 14, 200143.
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2017, 29, 61–74.
- Delli Bovi, A.P.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-Alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371.
- Mittermeier, C.; Konopa, A.; Muehlich, S. Molecular Mechanisms to Target Cellular Senescence in Hepatocellular Carcinoma. Cells 2020, 9, 2540.
- Aravinthan, A. Cellular Senescence: A Hitchhiker’s Guide. Hum. Cell 2015, 28, 51–64.
- Smith, J.; Mun Tho, L.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2010; Volume 108.
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827.
- D’Adda Di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198.
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118.
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular Senescence and the Senescent Secretory Phenotype: Therapeutic Opportunities. J. Clin. Investig. 2013, 123, 966–972.
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990.
- Huda, N.; Khambu, B.; Liu, G.; Nakatsumi, H.; Yan, S.; Chen, X.; Ma, M.; Dong, Z.; Nakayama, K.I.; Yin, X.-M. Senescence Connects Autophagy Deficiency to Inflammation and Tumor Progression in the Liver. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 333–355.
- Kudlova, N.; De Sanctis, J.B.; Hajduch, M. Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. Int. J. Mol. Sci. 2022, 23, 4168.
- Adewoye, A.B.; Tampakis, D.; Follenzi, A.; Stolzing, A. Multiparameter Flow Cytometric Detection and Quantification of Senescent Cells In Vitro. Biogerontology 2020, 21, 773–786.
- Itahana, K.; Campisi, J.; Dimri, G.P. Methods to Detect Biomarkers of Cellular Senescence: The Senescence-Associated Beta-Galactosidase Assay. Methods Mol. Biol. 2007, 371, 21–31.
- Ferreira-Gonzalez, S.; Rodrigo-Torres, D.; Gadd, V.L.; Forbes, S.J. Cellular Senescence in Liver Disease and Regeneration. Semin. Liver Dis. 2021, 41, 50–66.
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific Lipofuscin Staining as a Novel Biomarker to Detect Replicative and Stress-Induced Senescence. A Method Applicable in Cryo-Preserved and Archival Tissues. Aging 2013, 5, 37–50.
- Zhang, X.; Zhou, D.; Strakovsky, R.; Zhang, Y.; Pan, Y.X. Hepatic Cellular Senescence Pathway Genes Are Induced through Histone Modifications in a Diet-Induced Obese Rat Model. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, 558–564.
- Ping, F.; Li, Z.-Y.; Lv, K.; Zhou, M.-C.; Dong, Y.-X.; Sun, Q.; Li, Y.-X. Deoxyribonucleic Acid Telomere Length Shortening Can Predict the Incidence of Non-Alcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus. J. Diabetes Investig. 2017, 8, 174–180.
- Laish, I.; Mannasse-Green, B.; Hadary, R.; Biron-Shental, T.; Konikoff, F.M.; Amiel, A.; Kitay-Cohen, Y. Telomere Dysfunction in Non-alcoholic Fatty Liver Disease and Cryptogenic Cirrhosis. Cytogenet. Genome Res. 2016, 150, 93–99.
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Non-alcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. 2016, 11, 451–496.
- Sriram, S.; Yuan, C.; Chakraborty, S.; Tay, W.; Park, M.; Shabbir, A.; Toh, S.; Han, W.; Sugii, S. Oxidative Stress Mediates Depot-Specific Functional Differences of Human Adipose-Derived Stem Cells. Stem Cell Res. Ther. 2019, 10, 141.
- Ross, R.; Soni, S.; Houle, S. Negative Energy Balance Induced by Exercise or Diet: Effects on Visceral Adipose Tissue and Liver Fat. Nutrients 2020, 12, 891.
- Lohr, K.; Pachl, F.; Moghaddas Gholami, A.; Geillinger, K.E.; Daniel, H.; Kuster, B.; Klingenspor, M. Reduced Mitochondrial Mass and Function Add to Age-Related Susceptibility toward Diet-Induced Fatty Liver in C57BL/6J Mice. Physiol. Rep. 2016, 4, e12988.
- Kondo, Y.; Hasegawa, G.; Okada, H.; Senmaru, T.; Fukui, M.; Nakamura, N.; Sawada, M.; Kitawaki, J.; Okanoue, T.; Kishimoto, Y.; et al. Leprdb/Db Mice with Senescence Marker Protein-30 Knockout (Leprdb/DbSmp30Y/-) Exhibit Increases in Small Dense-LDL and Severe Fatty Liver Despite Being Fed a Standard Diet. PLoS ONE 2013, 8, e65698.
- Park, H.; Ishigami, A.; Shima, T.; Mizuno, M.; Maruyama, N.; Yamaguchi, K.; Mitsuyoshi, H.; Minami, M.; Yasui, K.; Itoh, Y.; et al. Hepatic Senescence Marker Protein-30 Is Involved in the Progression of Non-alcoholic Fatty Liver Disease. J. Gastroenterol. 2010, 45, 426–434.
- Meijnikman, A.S.; Herrema, H.; Scheithauer, T.P.M.; Kroon, J.; Nieuwdorp, M.; Groen, A.K. Evaluating Causality of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. JHEP Rep. 2021, 3, 100301.
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How Does Hepatic Lipid Accumulation Lead to Lipotoxicity in Non-Alcoholic Fatty Liver Disease? Hepatol. Int. 2021, 15, 21–35.
- Petrosillo, G.; Portincasa, P.; Grattagliano, I.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Ferri, D.; Paradies, G. Mitochondrial Dysfunction in Rat with Nonalcoholic Fatty Liver: Involvement of Complex I, Reactive Oxygen Species and Cardiolipin. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1260–1267.
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular Senescence Drives Age-Dependent Hepatic Steatosis. Nat. Commun. 2017, 8, 70–72.
- Zhang, J.; Shi, Y. In Search of the Holy Grail: Toward a Unified Hypothesis on Mitochondrial Dysfunction in Age-Related Diseases. Cells 2022, 11, 1906.
- Greten, T.F.; Eggert, T. Cellular Senescence Associated Immune Responses in Liver Cancer. Hepatic Oncol. 2017, 4, 123–127.
- Liu, P.; Tang, Q.; Chen, M.; Chen, W.; Lu, Y.; Liu, Z.; He, Z. Hepatocellular Senescence: Immunosurveillance and Future Senescence-Induced Therapy in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 589908.
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and Tumour Clearance Is Triggered by P53 Restoration in Murine Liver Carcinomas. Nature 2007, 445, 656–660.
- Gungor, M.Z.; Uysal, M.; Senturk, S. The Bright and the Dark Side of TGF-β Signaling in Hepatocellular Carcinoma: Mechanisms, Dysregulation, and Therapeutic Implications. Cancers 2022, 14, 940.
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming Growth Factor-Beta Induces Senescence in Hepatocellular Carcinoma Cells and Inhibits Tumor Growth. Hepatology 2010, 52, 966–974.
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547.
- Huang, Y.; Yang, X.; Meng, Y.; Shao, C.; Liao, J.; Li, F.; Li, R.; Jing, Y.; Huang, A. The Hepatic Senescence-Associated Secretory Phenotype Promotes Hepatocarcinogenesis through Bcl3-Dependent Activation of Macrophages. Cell Biosci. 2021, 11, 173.
- Donati, B.; Pietrelli, A.; Pingitore, P.; Dongiovanni, P.; Caddeo, A.; Walker, L.; Baselli, G.; Pelusi, S.; Rosso, C.; Vanni, E.; et al. Telomerase Reverse Transcriptase Germline Mutations and Hepatocellular Carcinoma in Patients with Non-alcoholic Fatty Liver Disease. Cancer Med. 2017, 6, 1930–1940.
- Kim, S.K.; Ueda, Y.; Hatano, E.; Kakiuchi, N.; Takeda, H.; Goto, T.; Shimizu, T.; Yoshida, K.; Ikura, Y.; Shiraishi, Y.; et al. TERT Promoter Mutations and Chromosome 8p Loss Are Characteristic of Non-alcoholic Fatty Liver Disease-Related Hepatocellular Carcinoma. Int. J. Cancer 2016, 2518, 2512–2518.
- Papatheodoridi, A.M.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Non-alcoholic Fatty Liver Disease and Progression to Non-alcoholic Steatohepatitis. Hepatology 2020, 71, 363–374.
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-Induced Gut Microbial Metabolite Promotes Liver Cancer through Senescence Secretome. Nature 2013, 499, 97–101.
- Cai, X.; Guillot, A.; Liu, H. Cellular Senescence in Hepatocellular Carcinoma: The Passenger or the Driver? Cells 2022, 12, 132.
- Wang, C.; Chen, W.J.; Wu, Y.F.; You, P.; Zheng, S.Y.; Liu, C.C.; Xiang, D.; Wang, M.J.; Cai, Y.C.; Zhao, Q.H.; et al. The Extent of Liver Injury Determines Hepatocyte Fate toward Senescence or Cancer Article. Cell Death Dis. 2018, 9, 575.

