1. Oxidative Stress and Liver Metabolic Disease
1.1. The Oxygen Paradox
The appearance of molecular oxygen (O
2) in the earth’s atmosphere approximately 2.1–2.4 billion years ago led to a change in biodiversity and evolutionary adaptations in living organisms’ metabolism, switching from anaerobic to aerobic
[1][22]. For all aerobic organisms, O
2 is a key molecule in oxidative energy production, acting as the terminal acceptor of electrons in the final step of the mitochondrial electron transport chain
[2][23]. However, its chemical properties allow for the generation of intermediate by-products of O
2 metabolism with oxidizing capacity themselves, which can negatively affect cells and, by extension, organisms
[2][23]. Thus, an inconvenient paradox is revealed; O
2 is indispensable for the survival and growth of aerobic organisms while, in parallel, it has deleterious effects on them
[3][24].
1.2. ROS in Various Biological Roles
The constellation of molecules that are produced via the incomplete reduction of O
2 are collectively known as reactive oxygen species (ROS). ROS is a collective term that includes free radicals (species that contain at least one unpaired valency electron), but also some non-radical derivatives of O
2. “Reactive“ is a relative term, as these molecules display varying oxidative capacity
[4][25]. On one side of the spectrum, molecules such as the superoxide anion (O
2•−) and hydrogen peroxide (H
2O
2) are relatively unreactive. On the other side of the spectrum, species such as the hydroxyl radical (•OH), which is generated via the non-enzymatic oxidation of H
2O
2, are extremely reactive and can oxidise in a non-discrete manner any biological macromolecule in its close vicinity
[2][4][23,25].
A characteristic example of cysteine-based structural changes linked to redox sensing is the Nrf2/KEAP1 system. Nrf2 (Nuclear factor E2 related factor 2) is a transcription factor that enables the expression of multiple antioxidant enzymes. Keap1 (Kelch-like ECH-associated protein 1) mediated ubiquitination of Nrf2 under unstressed conditions controls the expression of Nrf2 target genes. However, OS-mediated cysteine residue modifications in Keap1 render it unable to mediate Nrf2 ubiquitination, leading to antioxidant enzymes gene expression
[4][25].
1.3. ROS Regulation and Removal—Oxidative Eustress and Distress
During their evolutionary course, eukaryotic cells have developed fine-tuned defence mechanisms that can rapidly eliminate the continuously generated O
2-derived species
[5][28]. A complex and heterogenous assortment of antioxidant enzymes, synthesized by all known aerobic organisms, provide a first line of oxidative defence. As such, superoxide dismutase (SOD) catalyses the dismutation of O
2•− into O
2 and H
2O
2, while the latter can be safely reduced to H
2O by catalases (Cat), glutathione peroxidases (Gpx), and peroxiredoxins (Prx)
[4][6][25,29].
The ability of certain ROS to alter macromolecular structures within a cell or cellular compartment, causing structural damage and subsequent dysfunction, was described by Harman in 1956
[7][31]. In 1972 he expanded his theory: free radicals that are mainly generated in mitochondria as by-products of aerobic metabolism cause oxidative damage. The accumulated oxidative damage to essential macromolecules contributes to aging; the mitochondrial free radical theory of ageing was later renamed oxidative stress theory (OST)
[8][9][30,32]. The long-term accumulation of faulty macromolecules that cells cannot dispose of leads to disruptions in signalling pathways, as well as a self-perpetuating cycle of metabolic dysregulation and OS, resulting inadvertently in cellular ageing
[10][11][33,34].
1.4. The Role of Iron in OS
An element that is often implicated in redox reactions and oxidative stress regulation is iron. In the human organism, ferrous (Fe
2+) or ferric (Fe
3+) iron, either by participating in macromolecular configurations or freely available in small quantities, facilitates a diverse set of cellular functions such as oxygen transport in haemoglobin and myoglobin, cellular respiration, enzymatic reactions, and nucleotide metabolism. The involvement of iron in oxidative stress is classically illustrated in the Fenton reaction, where labile iron, which is normally freely available in the cell in minuscule amounts, catalyses the conversion of H
2O
2, a relatively weak ROS, to the •OH. As previously mentioned, the •OH is extremely reactive and can oxidise any macromolecule in its vicinity, leading to temporary—dependent on the available repair mechanisms—or permanent structural and functional damage
[1][12][22,35].
Furthermore, iron is indirectly implicated in H
2O
2-mediated signalling. This link is particularly demonstrated when iron chelation or depletion impacts adhesion molecule expression or transcriptional pathways modulation of lipopolysaccharide exposure response, linking iron with OS and inflammatory response
[1][22]. Labile iron modulation, either by iron-chelating drugs or by diet-derived iron-chelating compounds, was shown to play a key role in preventing H
2O
2-mediated apoptosis, and OS and iron metabolism are interconnected and tightly regulated
[13][14][36,37]. In Metabolic Associated Fatty Liver Disease (MAFLD), the dysmetabolic iron overload syndrome, a deranged iron metabolism phenotype, mostly evidenced as hepatocellular or reticuloendothelial iron overload, creates one of the prerequisites for redox imbalance and OS in the context of the disease
[15][6].
1.5. OS in Liver Metabolic Disease—The Concept of Lipotoxicity
For a deeper understanding of the role of OS in MS, one must consider the fact that some types of non-adipose cells, such as hepatocytes, β-cells, myocytes, and podocytes, display a certain capacity of storing lipids in their cytoplasm. Oversaturation of these cell types with lipids and their derivatives leads to a generalised metabolic dysfunction with multiple sequelae, termed lipotoxicity
[16][19]. In the case of MAFLD, the hepatocyte is the main site of lipotoxicity, and while the lipotoxic effects of fatty acids are mediated by multiple biological processes, of particular interest is OS.
The connection between OS and lipotoxicity can be traced to lipid peroxidation. Under OS conditions, polyunsaturated fatty acids can undergo peroxidation. The enzymatic pathway of lipid peroxidation is mediated by different enzymes, amongst which prominent is the role of iron-containing lipoxygenases (LOX), which produce lipid peroxides (LOOH)
[17][38]. Glutathione peroxidase 4 (Gxp4) is the key antioxidant enzyme in neutralising these lipid peroxides
[18][39].
When looking outside the hepatocyte, another aspect of MAFLD that implicates OS in its pathogenesis and disease progression is the altered gut microbiota. The changes in gut microbiota in MAFLD patients are attributed to the Western-type diet and insulin resistance. The resultant qualitative and quantitative changes in gut flora create an environment of dysbiosis that leads to an increased baseline inflammatory response of the resident immune cells, such as the hepatic Kupffer cells, which in turn show phenotypes of increased ROS release
[19][44].
2. Cellular Senescence
2.1. Overview of CS
CS, an irreversible state during which cells lose their ability to proliferate, is triggered by several endogenous and exogenous stimuli, such as aging, ROS, oncogene expression, and radiation
[20][45]. CS is a true example of the principle of antagonistic pleiotropy; when in younger individuals and precancerous or early-stage cancer, it serves as a tumour suppressive mechanism, whereas in older individuals or established cancer, its unique properties favour cancer cell proliferation.
There are two main mechanisms of CS, the replicative CS, which is induced normally under aging conditions, where shortening of telomeres leads inevitably to cell cycle arrest, and the stress-induced premature CS, where external or internal factors cause DNA damage, thereby inducing CS pathways independently of telomere length
[21][46]. Both mechanisms activate the same pathway, known as DNA damage response (DDR), which recruits Ataxia Telangiectasia Mutated (ATM) protein kinase to activate its target protein Checkpoint Kinase 2 (CHK2)
[22][23][47,48]. Subsequently, ATM and CHK2 stabilise p53 resulting in the upregulation of its target protein p21
[23][24][48,49]. Both p53 and p21 inhibit the phosphorylation of the Retinoblastoma 1 factor (RB1), which can now bind to the E2F transcription factor, subsequently preventing cell cycle progression
[23][48].
Apart from activating p53 and p16 to promote cell cycle arrest, DDR is also a well-known activator of senescence-associated secretory phenotype (SASP), thus senescent cells, despite entering a cell proliferation arrest, remain metabolically active and are able to communicate with their microenvironment
[23][48]. SASP is composed of several cytokines secreted from senescent cells, such as interleukins (IL-1b, IL-6, IL-8, ROS, growth factors, such as hepatocyte growth factor (HGF), and proteases, such as Matrix Metalloproteinases (MMPs)
[25][26][51,52]. SASP serves as a regulator of the cellular microenvironment, with autocrine and paracrine actions, by promoting immunological removal of senescent cells, reinforcing senescence in neighbouring cells, and paradoxically acquiring pro-tumorigenic properties
[27][53] depending on the stimulus that initially triggered CS
[28][54].
2.2. CS and Molecular Players
Although the detection and study of senescent cells are based on several morphological and biochemical features, none of them is completely indicative of CS; thus, a combination of characteristics is typically used to identify senescent cells
[29][55].
Despite the fact that several phenotypical changes have been observed in senescent cells, such as an enlarged and flattened shape, reorganisation of chromatin, and changes in the nuclear membrane
[30][56], these morphological features are not sufficient to detect CS. To that end, numerous studies have underlined specific biomarkers for the identification of senescent cells.
Proteins playing a crucial role in the induction of cell cycle arrest, such as p53/p21 and p16, are common senescence biomarkers
[29][55]. Furthermore, CS is often characterised by increased lysosomal activity, which can be detected by specific enzymes, such as senescence-associated β-galactosidase (SA-β-Gal), which constitutes a widely known senescence biomarker
[31][57]. Moreover, another established CS biomarker is lipofuscin, a yellowish-brown residue known as “age pigment”, consisting of incompletely degraded oxidized molecules due to increased lysosomal content
[32][58]. Interestingly, it has been demonstrated that SA-β-Gal co-localizes with lipofuscin; thus, these two molecules can be used alternatively to detect CS
[33][59]. An additional CS biomarker is the senescence-associated heterochromatin foci (SAHF), a group of heterochromatin and proteins that suppress the expression of proliferation genes
[34][60]. Some studies have also investigated the detection of SASP compounds, mainly associated with pro-inflammatory features of senescent cells; however, confirming CS based on these factors is still a matter of debate
[29][55].
3. MAFLD Specific Considerations
3.1. CS in MAFLD
There is substantial evidence indicating that CS, especially of the hepatocytes, may foster fat accumulation and inflammation in different stages of MAFLD
[32][58]. In this context, numerous studies have indulged in the role of CS in MAFLD progression, with major CS phenotypical characteristics, such as shorter telomeres and elevated CS biomarkers, such as p53 and SAHF, being observed in patients with MAFLD
[35][36][61,62].
An increased prevalence of MAFLD in older patients has set the context for further investigation of the role of CS in liver fat accumulation and propagation of the disease
[37][63]. However, as mentioned above, induction of CS is not only mediated by aging and telomere shortening, but also under stressful stimuli provoking DNA damage.
Given the complexity of hepatic architecture and function, as well as the broad phenotypic spectrum of MAFLD, one should consider how CS of other cell types helps establish MAFLD. The main regulator of CS in the hepatic microenvironment is hepatocyte-derived SASP, which depending on the duration of the pathological stimulus, can display a vast array of paracrine functions, activating, recruiting, or rendering senescent, adjacent, and distant cells.
3.2. CS and OS in MAFLD
The intertwined roles of OS and CS in extrahepatic MAFLD pathogenesis could not be better demonstrated than in the murine model of Sriram et al. Visceral adipose tissue-derived stem cells display high OS characteristics, with intense Nox activity linked to early CS phenotype, attenuated by ascorbic acid treatment, a potent antioxidant
[38][79]. Knowing the well-established connection between visceral adipose tissue and fatty liver disease, the above study shows the importance of extrahepatic CS in MAFLD
[39][80].
Lohr and colleagues also used a C57BL/6J mouse model to study the effects of a high-fat diet and age on MAFLD development. They analysed hepatic mitochondria and concluded that susceptibility to diet-induced obesity and fatty liver increased with age. This also correlated with an age-related reduction in mitochondrial mass and was aggravated by impaired fatty acid oxidation in high-fat diet mice
[40][85].
Kondo et al. examined Senescence Marker Protein–30 (SMP-30)—an antioxidant and anti-apoptotic protein that declines with increasing age—and observed that when comparing standard fat diet-fed mice with SMP-30 knockout to normal counterparts, the former developed fatty liver with inflammatory cells infiltrates and increased oxidative stress linked with impaired fatty acid oxidation and lipoprotein uptake
[41][86]. However, human hepatic biopsy studies of SMP-30 failed to draw conclusive results on the exact role of this ageing-related protein in MAFLD, despite the clear correlation between its expression and disease occurrence
[42][87].
3.3. Mitochondrial Dysfunction as a Link between OS and CS in MAFLD
Given the intense oxidative activity that normally takes place within the mitochondria, when the hepatocyte is oversaturated with fatty acids, as in the case of MAFLD, initially, β-oxidation is accelerated
[43][44][88,89]. When β-oxidation is intensified, mitochondrial ROS production increases dramatically. Fatty acids in the inner mitochondrial membrane are rapidly turned into lipid peroxides, intensifying the OS milieu and leading to macromolecular impairment (mitochondrial DNA, lipids, and proteins)
[45][90]. This functional impairment, in turn, deteriorates the already oversaturated β-oxidation system, leading to further OS deterioration, oxidative phosphorylation deficiency, and mitochondrial dysfunction-driven CS
[46][83]. A key player in mitochondrial dysfunction appears to be acyl-CoA:lysocardiolipin acyltransferase 1 (ALCAT1), an enzyme responsible for cardiolipin—a key structural mitochondrial membrane phospholipid–remodelling, activated under conditions of lipotoxicity and OS, whose detrimental impacts to the mitochondrial structure are mediated by mitofusin 2 (MFN2) downregulation
[45][47][90,91].
In total, all the above show a clear connection between OS-induced CS and the initiation and progression of MAFLD to NASH and fibrosis. High-fat hepatocyte concentrations, both dietary and from de novo lipogenesis—propagated by insulin resistance—impair hepatic lipid metabolism and generate high amounts of cytoplasmic and mitochondrial ROS, which in turn cause nuclear and mitochondrial DNA damage, triggering CS and installing an elaborate SASP. This SASP displays both autocrine functions, condemning the cell in a vicious circle of metabolic dysregulation secondary to deteriorating OS, and paracrine functions, remodelling the hepatic microenvironment, giving the microscopic characteristics of the MAFLD spectrum.
4. Does CS Protect from or Induce Hepatocarcinogenesis in the Context of MAFLD?
The role of CS in the progression of hepatocarcinogenesis is considered pivotal, as it has been shown to have both protective and tumorigenic properties.
CS is widely considered a potent tumour suppressive mechanism since the induction of senescence in premalignant hepatocytes has been shown to limit cancer development. One of the main features of senescent cells that allow for the early restriction of hepatic tumorigenesis is their capacity to control their surroundings via SASP secretion. Premalignant or malignant hepatocytes entering the CS programme secrete SASP factors that activate innate immunity and recruit local and distant macrophages to mediate their own clearance, thus halting the progression to an organized hepatocellular carcinoma (HCC) tumour. This mechanism of immune-mediated clearance of senescent cells is widely known as immunosurveillance and is of paramount importance as a barrier to hepatocarcinogenesis
[48][49][50][98,99,100]. Several SASP components have been studied, considering their suppressive role in HCC development. More specifically, TGF-β1 has been demonstrated to act in an autocrine manner by inducing ROS generation and perpetuating senescent phenotype in HCC mice cells, thereby leading to a restriction in tumour growth by 75%
[51][52][101,102].
Despite its repeatedly proven cancer-protective properties, when the hepatic micro-environment is changed, and tumour conditions are developed, CS can, under specific circumstances, usher HCC development. This has been explicitly shown in a series of experiments by Eggert et al., who demonstrated that hepatocyte senescence in tumour-free mice led to the recruitment of immune cells, a process mediated by SASP components, thus resulting in clearance of senescent hepatocytes, a well-known tumour repressing effect
[53][103]. However, when HCC cells were infused in mice with senescent hepatocytes, tumour growth was observed, and further analysis showed that the SASP-mediated immune cells inhibited NK and CD8+ T cells from clearing the cancerous hepatocytes
[53][103]. Another clear example of how the shifts in immunosurveillance can herald hepato-carcinogenesis was shown in the experimental and clinical work of Huang et al. Hepato-cellular CS induced a SASP characterized by IL-6 and IL-8 molecules, with macrophage activation. These macrophages displayed M2 polarization, a characteristic of advanced hepatocarcinogenesis, with pro-inflammatory and fibrotic properties, whereas they did not demonstrate any phagocytotic activity, as M1 macrophages show in early HCC, where they clear away premalignant senescent hepatocytes
[54][104].
A progressive shift to a quantitative increase in hepatocyte senescent phenotype driven by telomeres was observed by Donati et al.
[55][105] when comparing cohorts of MAFLD cirrhotic to HCC patients. They showed a higher prevalence of germline mutations of the telomerase reverse transcriptase (TERT), associated with reduced telomere length in HCC patients versus their cirrhotic counterparts
[55][105]. The role of telomere attrition secondary to TERT dysfunction in early MAFLD versus other aetiologies of chronic liver disease has also been highlighted in other studies
[36][62]. Despite these two studies showing a linear relationship between replicative senescence and the natural history of MAFLD, spontaneous somatic mutations of any of the telomerase components due to oxidative stress can trigger a CS-phenotype and act as a possible mechanism of MAFLD-associated hepatocarcinogenesis. Last, TERT promoter somatic mutations along with 8p loss were shown to be characteristic genomic changes of HCC secondary to MAFLD, frequently accompanied by TP53 as well as SWI/SNF complex constituents (ARID1A and ARID2) mutations, clarifying the landscape of genomic instability in these patients
[56][106]. Thus, it is evident that hepatocellular senescence, when maintained in an advanced HCC milieu, can harbour hepatocarcinogenesis by altering SASP and immune cell signalling.
In addition to the role of senescent hepatocytes in HCC development, it is also worth considering the impact of CS in other cellular populations and its effects on tumour biology. Intriguingly, it is well established that the development of HCC secondary to MAFLD often occurs in the absence of liver cirrhosis or in livers with a low degree of fibrosis
[15][6]. In this context, studies have shown that MAFLD-associated factors, such as obesity, have been linked with hepatic stellate cells (HSCs) senescence and expression of tumour-promoting SASP
[57][21]. Although senescence of HSCs has been considered as a protective mechanism against fibrosis and subsequently to HCC, this seems to be reversed in the case of steatohepatic HCC, as depletion of senescent HSCs prevented HCC development in obese mice
[58][107].
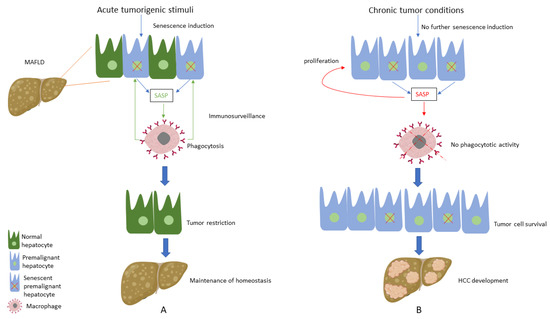
Considering the above-mentioned pivotal role of CS regarding hepatocarcinogenesis, studies have focused on the factors that trigger this alteration of the CS profile. Interestingly, chronic moderate liver injury, similar to the one MAFLD causes to the liver, led to the development of HCC, while the induction of acute injury in a chronic injury environment activated hepatocellular senescence and subsequently caused HCC reduction
[59][108], showing the detrimental impact of chronic low-intensity stressors in hepatic tumorigenesis. In this context,
thwe
researchers ssuppose that hepatocytes sense initial premalignant stimuli as an acute liver injury during the early stages of HCC development; thus, induction of senescence in premalignant hepatocytes acts as a protective mechanism, leading to the clearance of premalignant cells and tumour restriction
[60][109]. On the other hand, when tumour conditions are established following chronic moderate MAFLD-associated tumorigenic stimuli, senescence is not further activated; thus, premalignant hepatocytes do not enter this cell-cycle arrest process, thereby leading to HCC development and expansion. Furthermore, there is substantial evidence supporting that the existing senescent hepatocytes, instead of promoting immunosurveillance, seem to induce aberrant proliferation of the adjacent hepatocytes via SASP secretion; thus, abnormal proliferation of hepatocytes favours hepatic carcinogenesis
[59][108] (
Figure 1).
Figure 1. (A) Under acute tumorigenic conditions, activation of oncogenes and subsequent DNA damage response results in senescence induction, with senescent hepatocytes secreting multiple types of soluble factors, known as senescence-associated secretory phenotype (SASP). SASP results in the recruitment of immune cells, which eliminate premalignant senescent hepatocytes, a process known as immunosurveillance. Thus, under these conditions, senescence plays a protective role by restricting tumour development and contributing to the maintenance of homeostasis. (B) When tumorigenic stimuli become chronic, there is no further senescence induction in hepatocytes. SASP secreted from the existing senescent hepatocytes presents an altered profile by recruiting immune cells, which present no phagocytotic activity, while concomitantly contributing to the proliferation of premalignant cells. Thus, these conditions result in tumour cell survival and the development of hepatocellular carcinoma (HCC).