 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kamla Pathak | -- | 3996 | 2023-07-01 15:52:54 | | | |
| 2 | Peter Tang | Meta information modification | 3996 | 2023-07-03 05:03:05 | | |
Video Upload Options
The application of metallic nanoparticles as a novel therapeutic tool has significant potential to facilitate the treatment and diagnosis of mitochondria-based disorders. Subcellular mitochondria have been trialed to cure pathologies that depend on their dysfunction. Nanoparticles made from metals and their oxides (including gold, iron, silver, platinum, zinc oxide, and titanium dioxide) have unique modi operandi that can competently rectify mitochondrial disorders.
1. Introduction
2. Mitochondrial Dysfunction and Diseases

|
Disease |
Mitochondrial Abnormalities |
Management |
Ref. |
|---|---|---|---|
|
Cardiovascular diseases |
Impaired mitochondrial electron transport chain due to elevated LDL, greater ROS production disturbed cardiac tension, altered cytosolic Ca2+ flux, ischemia-reperfusion injury, and other diseases (such as diabetes mellitus) |
Control fatty acids and cholesterol level. Regulate enzymes i.e., mitochondrial creatin phosphate, creatin kinase, and ATP synthase. Modulate Ca2+ concentration in myocardium. |
[8] |
|
Diabetes |
MtDNA mutation Delayed Electron transport chain Increased beta oxidation and lipid accumulation Higher ROS overproduction Disturbed insulin signal pathway Increased intracellular glucose content, and constrained metabolism of glucose |
Prevention of ROS and lipid peroxidation. Reverse MtDNA change. Enhance glucose metabolism by inhibiting acetyl-CoA in mitochondria. |
[9] |
|
Kidney |
Mitochondrial DNA mutation (complex I–V) Hypomagnesemia Hypokalemia Hypoparathyroidasim Uremic toxins Kidney diseases |
Manage Erythropoetin signaling for normal mitochondrial biogenesis and metabolism. Hypoxia-inducible factor prolyl hydroxylase (HIF-PH) inhibitors, Nrf2-activating triterpenoid, sodium–glucose transporter 2 (SGLT2) inhibitors, and control of carnitine level |
[10] |
|
Alzheimer’s disease |
Aggregated Aβ peptide bond with a component that controls mitochondrial permeability ‘cyclophilin D’ Resultant reduction in membrane potential due to opening of pores. Free energy and ROS generated by beta amyloid peptide. Leads to MtDNA mutation and neuronal toxicity. |
Inhibition ofAβ peptide clusters binding with cyclophilin D. Restoration of MtDNA and enzyme replacement Inhibit cytochrome c and caspase activity Decrease mitochondrial fission |
[11] |
|
Cancer |
Enhanced mitochondrial complex I activity. Mutations in oncogenes Expression of oncoproteins (IDH 1 and 2, SDH and FH) Apoptosis induced factors released. Defective oxidative phosphorylation Hypoxic milieu proliferate condition |
Isocitrate dehydrogenase (IDH) 1 and 2 inhibitors Oxidative phosphorylation suppression Antioxidants Mitochondrial complex I and V inhibitors Lactate dehydrogenase (LDH) inhibitors |
[12] |
3. Therapeutic Approaches
|
Therapeutics |
Approach |
Model |
Outcome |
Ref. |
|---|---|---|---|---|
|
Upsurging ATP levels |
||||
|
Inosine and Febuxostat |
To increase ATP level and hypoxanthine in peripheral blood |
Patients with homoplasmic and heteroplasmic mutations |
After oral administration, brain natriuretic peptide (specific marker for heart failure) was reduced up to 31%. Moreover, 3.1-fold insulinogenic index was improved, suggesting a promising action of the given treatment. |
[13] |
|
Stimulating mitochondrial biogenesis |
||||
|
Bezafibrate and AMPK agonist 5-aminoimidazole-4-carboxamide ribonucleotide |
To initiate biogenesis and activate AMP protein kinase/PGC-1α-dependent pathway |
Double recombinant mice overexpressing PGC-1α in skeletal muscles |
Stimulation of PPAR/AMPK/PGC-1 alpha increased mitochondrial biogenesis, which monitors the homeostatic pathway and motor improvement in Sco2(KO/KI) animal model |
[14] |
|
5-Aminoimidazole-4-carboxamide ribotide (AICAR) |
ATP content and mitochondrial growth were aimed without disturbing membrane potential |
CI deficient fibroblasts, such as NDUFS2 and C20ORF7 |
Fluorescence microscopy detailed the activation of AMP protein kinase with AICAR |
[15] |
|
Nicotinamide riboside (NAD+ precursor) |
Effect of nicotinamide in pharmacokinetic parameters and NAD+ level in blood for the treatment of genetic or acquired mitochondrial abnormalities |
Impaired mitochondrial murine model |
Orally administered nicotinamide riboside was well tolerated, and an increased mean steady state concentration (Css, p = 0.03) two times greater than the baseline NAD+ concentration in blood was observed, whereas average circulating level of NAD+ at day 1 was 27 ± 6 µM. |
[16] |
|
Modulation of the Nitric acid or cGMP/PKG pathway |
||||
|
L-arginine (Nitric acid precursor) |
To reduce capacity for nitric acid-based vasodilation |
MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes) patients |
Obtained data suggested that prepared L-Arg infusion was most effective when administered within 4 hrs of the onset of brain disorder symptoms in the acute phase of MELAS sufferers |
[17] |
|
Neural progenitor cells (NPC) |
To preserve parental mtDNA and show metabolic shift toward oxidative phosphorylation |
Homoplasmic mutation in the mitochondrial gene (MT-ATP6) |
Human induced pluripotent stem cells originated NPC, providing a potential tool for mtDNA targeted drug screening to avoid nervous system disorder. |
[18] |
|
Antioxidant therapy |
||||
|
N-acetyl cysteine |
Supplementation with N-acetyl cysteine to modify the mitochondrial respiratory chain function |
MELAS patients containing common mutation, i.e., m.3243A>G, m.8344A>G |
Supplementation with N-acetyl cysteine improved 2-thiomodificationof tRNA, thus regulating protein synthesis. |
[19] |
|
Cysteamine bitartrate |
Enhancement of glutathione biosynthesis for the reduction of oxidative stress associated with mitochondrial diseases |
Zebrafish model and Caenorhabditis elegans model carrying Complex I defect |
Cysteamine bitartrate improved mitochondrial membrane potential in nephropathic cystinosis and cured multiple RC complex diseases in FBXL4 human at 10 to 100 μm concentrations. |
[20] |
|
Inclusion of Redox-Active Molecules |
||||
|
Tc99m-HMPAO |
To explore Tc99m-HMPAO for determination of glutathione/protein thiol levels in cerebral blood flow |
Pediatric mitochondrial diseased patients |
The patients showed improvement in Newcastle score (n = 5, p = 0.028), confirming the potential of Tc99m-HMPAO as a bioimaging marker for the oxidative state of brain. |
[21] |
|
EPI-743 |
Management of cellular oxidative stress in mitochondrial respiratory chain disease |
5-month-old girl suffering from Leigh syndrome |
EPI-743 (a potent stress protectant) exhibited improvement in mitochondrial associated issues, i.e., improvement in eye, motor, and bowel movements |
[22] |
|
Monitoring of mitochondrial dynamics |
||||
|
Cytotoxic Necrotizing Factor-1 |
To control mitochondrial impairment and cell damage |
Patient sufferer from m.8344A>G gene |
CNF-1 triggered energetic content of mitochondria via activation of actin cycloskeleton in Myoclinic epilepsy with ragged red fibers (MERRF) and increased mitochondrial marker tomo20 |
[23] |
|
Modulating mitochondrial autophagy |
||||
|
Rapamycin |
Inhibition of mTOR in mitochondrial defect in Leigh syndrome |
Ndufs4 wild type mouse |
Rapamycin slowed down neurological symptoms and minimized neuroinflammation in brain lesions. It also slowed the formation of glycolytic intermediates. |
[24] |
|
NADH dehydrogenase Ndi1 |
To replace mitochondrial complex 1 for reoxidation of NADH intramitochondrially. |
Transgenic strains of Drosophila |
Overexpression of NDI1 relieves aging and manages production of ROS. Deposition of these oxidative damaged markers is declined in aged flies. |
[25] |
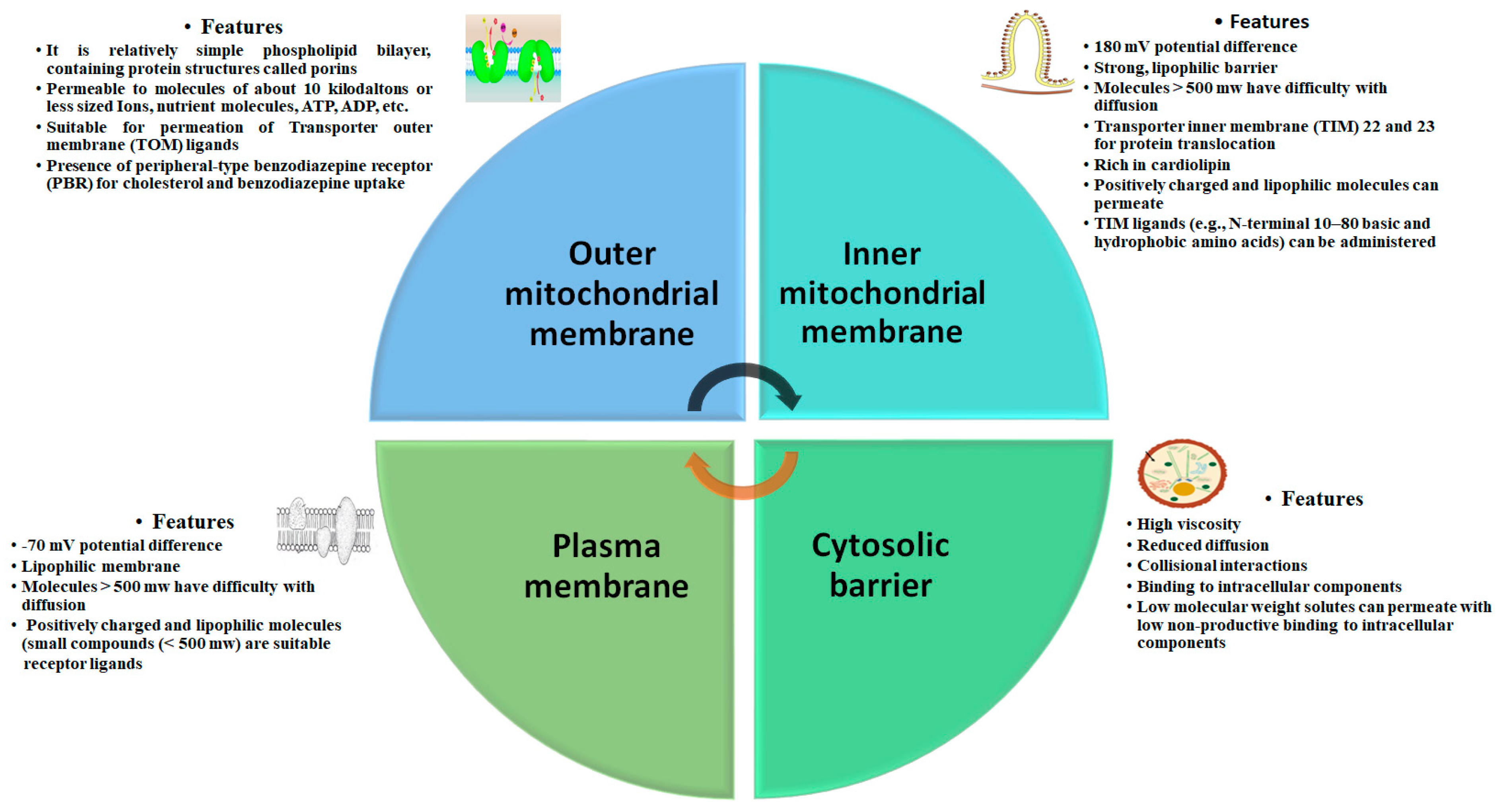
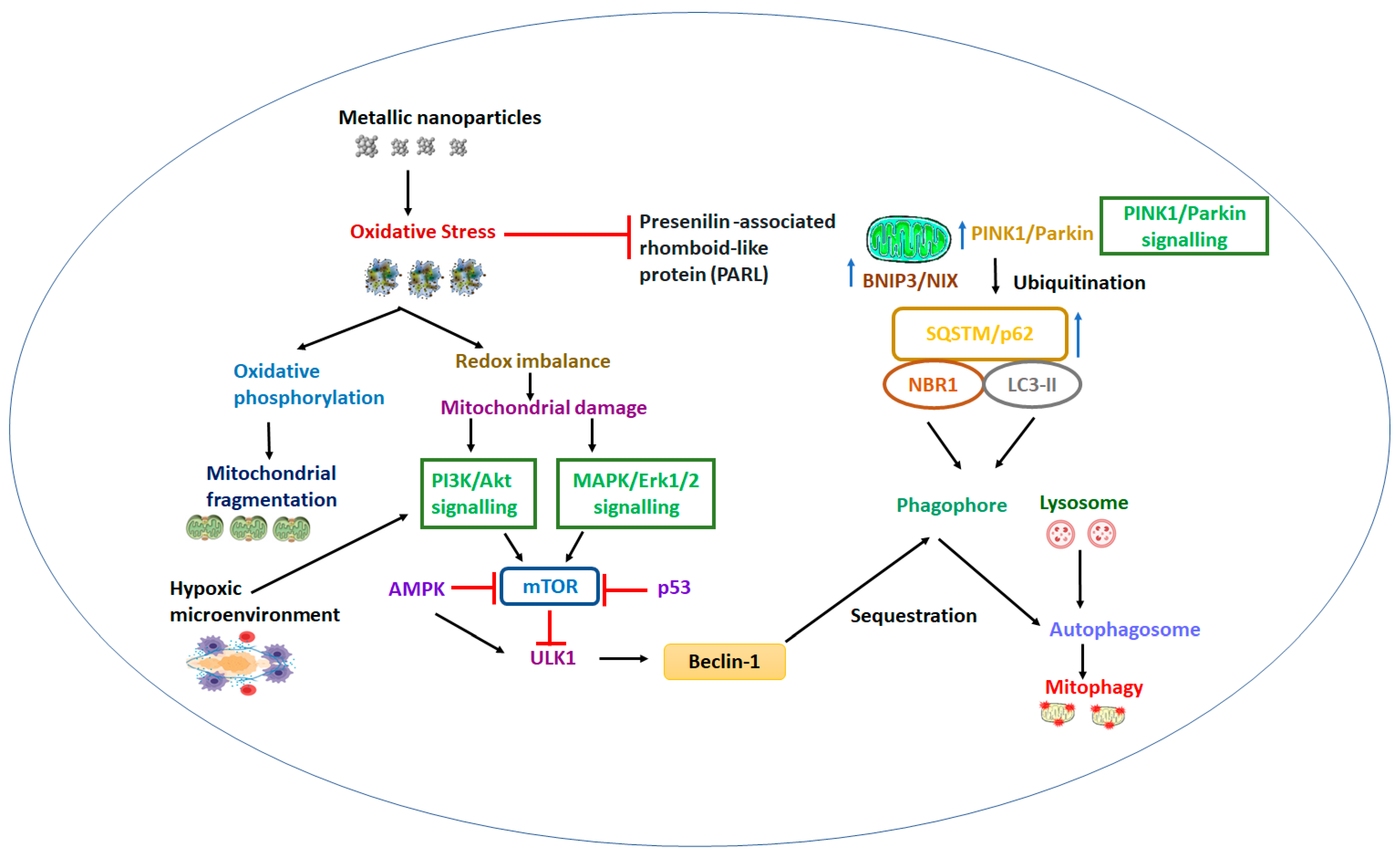
4. Biological Barrier and Toxicity
5. Nanoengineered Mitochondria Targeted Approaches
|
Nanodrug Delivery System |
Purpose |
Model |
Outcomes |
Relevance |
Ref. |
|---|---|---|---|---|---|
|
Topotecan loaded liposome |
Mitochondrial targeted system to overcome resistant related metastases |
Multi drug Resistant MCF-7/ADR cell xenografts |
Mitochondrial targeted liposomes were 64.84 nm with −0.52 ± 0.08 mV membrane potential. Encapsulation efficiency was ≥95%. They were stable in physiological blood system and exhibited minimal leakage. The system led to release cytochrome C, stimulated caspase 9 and 3, and exhibited superior inhibitory action on the resistant B16 melanoma metastatic mice. |
Topotecan localized in mitochondria that exhibited potent inhibitory action on the on the resistant B16 metastatic melanoma. |
[44] |
|
Paclitaxel loaded triphenylphosphine nanomicelles |
Inhibition of antiapoptotic Bcl-2 |
Drug-resistant breast cancer-bearing mouse model with lung metastasis (A549/ADRcells) |
The nanomicelles were small (142 ± 8.35 nm) with PDI 0.235 and negative zeta potential (−24.65 ). This system significantly hampered A549/ADR cells and deposited over mitochondria surface. Inhibition of Bcl-2 led to release cytochrome C and triggered caspase 3 and 9, mediating mitochondrial outer membrane permeabilization. |
Positively charged nanomicelles adhered inside mitochondria and exhibited apparent multidrug resistant tumor targeting efficacy |
[45] |
|
Curcumin loaded polymeric and lipid nanosuspensions |
To neutralize generation of reactive oxygen species due to disturbance of signal proteins in mitochondria. |
Olfactory ensheathing cells |
Uniform and spherical polymeric and lipid nanosuspensions were of mean sizes 338 nm and 127 nm, respectively, with negatively charged zeta potential. The nanosuspensions were quite stable for more than 135 days. Improved cell viability suggested potential incorporation of curcumin in nanosuspension against hypoxic cells of Olfactory ensheathing cells at the concentration of 5 µM. |
Potential intranasal polymeric and lipid nanosuspention was advised for neuroprotective action due to antioxidant property of curcumin. |
[46] |
|
Hydroxyl-terminated polyamidoamine (PAMAM)-N-acetyl cysteine dendrimers |
Mitochondrial targeted delivery in oxidative stress-induced glial cell |
Rabbit traumatic brain injury (TBI) model |
Significantly high localization of drug in mitochondria than nonmodified dendrimer due to potential for attenuation of oxidative stress. Systemic administration in TBI model of rabbit exhibited capability to penetrate BBB and target glial cells due to localization in the white matter of the injured hemisphere. |
Colocalization of dendrimer in mitochondria of glial cells in traumatic brain injury |
[47] |
|
Osthole nanoemulsion (OST-NE) |
Regulation of apoptosis pathway and mitochondrial oxidative stress. |
Alzheimer’s disease model mice |
Intranasal delivery of osthole nanoemulsion (mean particle size 2.33 nm) enhanced bioavailability, regulated cholinergic system, maintained mitochondrial potential, and inhibited apoptosis and oxidative stress. |
OST-NE lessened upstream modulator Bax, that depolarized mitochondrial membrane potentail and exhibited antiapoptotic effect or neuroprotective action in Alzheimer disease |
[48] |
|
Momordica charantia silver nanoparticles |
Uphold mitochondria biogenesis and enhanced the expression of PPARϒ, an energy metabolism coordinator |
Pancrease of diabetic rats |
Silver nanoparticles possessed irregular/uneven surface and diameter. Contained Momordica charantia enhanced glucose sensitivity in the dibetic rat model at a lower dose of 50 mg/kg by slowing down JAK/STAT and AKT/PI3K pathways in mitochondria. |
Developed nanoparticles promoted glucose uptake and insulin secretion via improving mitochondrial biogenesis in pancreas of diabetic rats. |
[49] |
|
Dequalinium embedded DQAsomes |
Dicationic amphiphilic was investigated for affinity towards binding with mitochondria DNA. |
Plasmid DNA firefly luciferase |
Developed DQAsomes created a liposome-like aggregate system in aqueous media that were able to bind with DNA and had efficiency to transfect cells compared to Lipofectin™ reagent. |
DQAsomes selectively accumulated in mitochondria of cancerous cells, and suggeted potential use as a nonviral transfection vector in gene delivery system. |
[50] |
References
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646.
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89.
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genom. Hum. Genet. 2010, 11, 25–44.
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569.
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age-related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184.
- Calabrese, V.; Lodi, R.; Tonon, C. D’Agata. V.; Sapienza, M.; Scapagnini, G.; Mangiameli, A.; Pennisi, G.; Stella, A.M.; Butterfield, D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci. 2005, 233, 145–162.
- Hellebrekers, D.M.; Wolfe, R.; Hendrickx, A.T.; de Coo, I.F.; de Die, C.E.; Geraedts, J.P.; Chinnery, P.F.; Smeets, H.J. PGD and heteroplasmic mitochondrial DNA point mutations: A systematic review estimating the chance of healthy offspring. Hum. Reprod. Update 2012, 18, 341–349.
- Bisaccia, G.; Ricci, F.; Gallina, S.; Di Baldassarre, A.; Ghinassi, B. Mitochondrial Dysfunction and Heart Disease: Critical Appraisal of an Overlooked Association. Int. J. Mol. Sci. 2021, 22, 614.
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532.
- Takemura, K.; Nishi, H.; Inagi, R. Mitochondrial Dysfunction in Kidney Disease and Uremic Sarcopenia. Front. Physiol. 2020, 11, 565023.
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588.
- Liu, Y.; Shi, Y. Mitochondria as a target in cancer treatment. MedComm 2020, 1, 129–139.
- Kamatani, N.; Kushiyama, A.; Toyo-Oka, L.; Toyo-Oka, T. Treatment of two mitochondrial disease patients with a combination of febuxostat and inosine that enhances cellular ATP. J. Hum. Genet. 2019, 64, 351–353.
- Viscomi, C.; Bottani, E.; Civiletto, G.; Cerutti, R.; Moggio, M.; Fagiolari, G.; Schon, E.A.; Lamperti, C.; Zeviani, M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell. Metab. 2011, 14, 80–90.
- Golubitzky, A.; Dan, P.; Weissman, S.; Link, G.; Wikstrom, J.D.; Saada, A. Screening for active small molecules in mitochondrial complex I deficient patient’s fibroblasts, reveals AICAR as the most beneficial compound. PLoS ONE 2011, 6, e26883.
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Nagana Gowda, G.A.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459.
- Sudo, A.; Sano, H.; Kawamura, N. Determination of the critical time point for efficacy of L-arginine infusion therapy in a case of MELAS with frequent stroke-like episodes. No Hattatsu 2014, 46, 39–43.
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Auré, K.; et al. Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell 2017, 20, 659–674.e9.
- Bartsakoulia, M.; Mϋller, J.S.; Gomez-Duran, A.; Yu-Wai-Man, P.; Boczonadi, V.; Horvath, R. Cysteine Supplementation May be Beneficial in a Subgroup of Mitochondrial Translation Deficiencies. J. Neuromuscul. Dis. 2016, 3, 363–379.
- Guha, S.; Konkwo, C.; Lavorato, M.; Mathew, N.D.; Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Polyak, E.; Lightfoot, R.; Seiler, C.; et al. Pre-clinical evaluation of cysteamine bitartrate as a therapeutic agent for mitochondrial respiratory chain disease. Hum. Mol. Genet. 2019, 28, 1837–1852.
- Blankenberg, F.G.; Kinsman, S.L.; Cohen, B.H.; Goris, M.L.; Spicer, K.M.; Perlman, S.L.; Krane, E.J.; Kheifets, V.; Thoolen, M.; Miller, G.; et al. Brain uptake of Tc99m-HMPAO correlates with clinical response to the novel redox modulating agent EPI-743 in patients with mitochondrial disease. Mol. Genet. Metab. 2012, 107, 690–699.
- Kouga, T.; Takagi, M.; Miyauchi, A.; Shimbo, H.; Iai, M.; Yamashita, S.; Murayama, K.; Klein, M.B.; Miller, G.; Goto, T.; et al. Japanese Leigh syndrome case treated with EPI-743. Brain Dev. 2018, 40, 145–149.
- Fabbri, A.; Travaglione, S.; Maroccia, Z.; Guidotti, M.; Pierri, C.L.; Primiano, G.; Servidei, S.; Loizzo, S.; Fiorentini, C. The Bacterial Protein CNF1 as a Potential Therapeutic Strategy against Mitochondrial Diseases: A Pilot Study. Int. J. Mol. Sci. 2018, 19, 1825.
- Johnson, S.C.; Yanos, M.E.; Kayser, E.B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 2013, 342, 1524–1528.
- Sanz, A.; Soikkeli, M.; Portero-Otín, M.; Wilson, A.; Kemppainen, E.; McIlroy, G.; Ellilä, S.; Kemppainen, K.K.; Tuomela, T.; Lakanmaa, M.; et al. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc. Natl. Acad. Sci. USA 2010, 107, 9105–9110.
- Burkhardt, C.; Kelly, J.P.; Lim, Y.H.; Filley, C.M.; Parker, W.D., Jr. Neuroleptic medications inhibit complex I of the electron transport chain. Ann. Neurol. 1993, 33, 512–517.
- Pathak, R.K.; Kolishetti, N.; Dhar, S. Targeted nanoparticles in mitochondrial medicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 315–329.
- Yu, H.; Koilkonda, R.D.; Chou, T.-H.; Porciatti, V.; Ozdemir, S.S.; Chiodo, V.; Boye, S.L.; Boye, S.E.; Hauswirth, W.W.; Lewin, A.S. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, E1238–E1247.
- Kagan, V.E.; Borisenko, G.G.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Potapovich, A.I.; Kini, V.; Amoscato, A.A.; Fujii, Y. Oxidative lipidomics of apoptosis: Redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free. Radic. Biol. Med. 2004, 37, 1963–1985.
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31.
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418.
- Will, Y.; Shields, J.E.; Wallace, K.B. Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology 2019, 8, 32.
- Nadanaciva, S.; Dykens, J.A.; Bernal, A.; Capaldi, R.A.; Will, Y. Mitochondrial impairment by PPAR agonists and statins identified via immunocaptured OXPHOS complex activities and respiration. Toxicol. Appl. Pharmacol. 2007, 223, 277–287.
- Dykens, J.A.; Jamieson, J.D.; Marroquin, L.D.; Nadanaciva, S.; Xu, J.J.; Dunn, M.C.; Smith, A.R.; Will, Y. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol. Sci. 2008, 103, 335–345.
- Dykens, J.A.; Jamieson, J.; Marroquin, L.; Nadanaciva, S.; Billis, P.A.; Will, Y. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically poised HepG2 cells and human hepatocytes in vitro. Toxicol. Appl. Pharmacol. 2008, 233, 203–210.
- Raza, H.; John, A. Implications of altered glutathione metabolism in aspirin-induced oxidative stress and mitochondrial dysfunction in HepG2 cells. PLoS ONE 2012, 7, e36325.
- Bonifacio, A.; Mullen, P.J.; Mityko, I.S.; Navegantes, L.C.; Bouitbir, J.; Krähenbühl, S. Simvastatin induces mitochondrial dysfunction and increased atrogin-1 expression in H9c2 cardiomyocytes and mice in vivo. Arch. Toxicol. 2016, 90, 203–215.
- Marrache, S.; Pathak, R.K.; Darley, K.L.; Choi, J.H.; Zaver, D.; Kolishetti, N.; Dhar, S. Nanocarriers for tracking and treating diseases. Curr. Med. Chem. 2013, 20, 3500–3514.
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20.
- Marrache, S.; Dhar, S. Engineering of blended nanoparticle platform for delivery of mitochondriaacting therapeutics. Proc. Natl. Acad. Sci. USA 2012, 109, 16288–16293.
- Bae, Y.; Jung, M.K.; Lee, S.; Song, S.J.; Mun, J.Y.; Green, E.S.; Han, J.; Ko, K.S.; Choi, J.S. Dequalinium-based functional nanosomes show increased mitochondria targeting and anticancer effect. Eur. J. Pharm. Biopharm. 2018, 124, 104–115.
- Vaidya, B.; Paliwal, R.; Rai, S.; Khatri, K.; Goyal, A.K.; Mishra, N.; Vyas, S.P. Cell-selective mitochondrial targeting: A new approach for cancer therapy. Cancer Ther. 2009, 7, 141–148.
- Choi, Y.S.; Cho, T.S.; Kim, J.M.; Han, S.W.; Kim, S.K. Amine terminated G-6 PAMAM dendrimer and its interaction with DNA probed by Hoechst 33258. Biophys. Chem. 2006, 121, 142–149.
- Yu, Y.; Wang, Z.H.; Zhang, L.; Yao, H.J.; Zhang, Y.; Li, R.J.; Ju, R.J.; Wang, X.X.; Zhou, J.; Li, N.; et al. Mitochondrial targeting topotecan-loaded liposomes for treating drug-resistant breast cancer and inhibiting invasive metastases of melanoma. Biomaterials 2012, 33, 1808–1820.
- Wang, H.; Zhang, F.; Wen, H.; Shi, W.; Huang, Q.; Huang, Y.; Xie, J.; Li, P.; Chen, J.; Qin, L.; et al. Tumor- and mitochondria-targeted nanoparticles eradicate drug resistant lung cancer through mitochondrial pathway of apoptosis. J. Nanobiotechnol. 2020, 18, 8.
- Bonaccorso, A.; Pellitteri, R.; Ruozi, B.; Puglia, C.; Santonocito, D.; Pignatello, R.; Musumeci, T. Curcumin Loaded Polymeric vs. Lipid Nanoparticles: Antioxidant Effect on Normal and Hypoxic Olfactory Ensheathing Cells. Nanomaterials 2021, 11, 159.
- Sharma, A.; Liaw, K.; Sharma, R.; Zhang, Z.; Kannan, S.; Kannan, R.M. Targeting Mitochondrial Dysfunction and Oxidative Stress in Activated Microglia using Dendrimer-Based Therapeutics. Theranostics 2018, 8, 5529–5547.
- Song, Y.; Wang, X.; Wang, X.; Wang, J.; Hao, Q.; Hao, J.; Hou, X. Osthole-Loaded Nanoemulsion Enhances Brain Target in the Treatment of Alzheimer’s Disease via Intranasal Administration. Oxidative Med. Cell. Longev. 2021, 2021, 8844455.
- Elekofehinti, O.O.; Ayodele, O.C.; Iwaloye, O. Momordica charantia nanoparticles promote mitochondria biogenesis in the pancreas of diabetic-induced rats: Gene expression study. Egypt. J. Med. Hum. Genet. 2021, 22, 80.
- Weissig, V.; Lasch, J.; Erdos, G.; Meyer, H.W.; Rowe, T.C.; Hughes, J. DQAsomes: A novel potential drug and gene delivery system made from Dequalinium. Pharm. Res. 1998, 15, 334–337.
- Wang, L.; Liu, Y.; Li, W.; Jiang, X.; Ji, Y.; Wu, X.; Xu, L.; Qiu, Y.; Zhao, K.; Wei, T.; et al. Selective targeting of gold nanorods at the mitochondria of cancer cells: Implications for cancer therapy. Nano Lett. 2011, 11, 772–780.
- Kreyling, W.G.; Semmler-Behnke, M.; Seitz, J.; Scymczak, W.; Wenk, A.; Mayer, P.; Takenaka, S.; Oberdörster, G. Size dependence of the translocation of inhaled iridium and carbon nanoparticle aggregates from the lung of rats to the blood and secondary target organs. Inhal. Toxicol. 2009, 21, 55–60.
- Recordati, C.; De Maglie, M.; Bianchessi, S.; Argentiere, S.; Cella, C.; Mattiello, S.; Cubadda, F.; Aureli, F.; D’Amato, M.; Raggi, A.; et al. Tissue distribution and acute toxicity of silver after single intravenous administration in mice: Nano-specific and size-dependent effects. Part. Fibre Toxicol. 2016, 13, 12.
- Zoroddu, M.A.; Medici, S.; Ledda, A.; Nurchi, V.M.; Lachowicz, J.I.; Peana, M. Toxicity of nanoparticles. Curr. Med. Chem. 2014, 21, 3837–3853.
- Paek, H.J.; Lee, Y.J.; Chung, H.E.; Yoo, N.H.; Lee, J.A.; Kim, M.K.; Lee, J.K.; Jeong, J.; Choi, S.J. Modulation of the pharmacokinetics of zinc oxide nanoparticles and their fates in vivo. Nanoscale 2013, 5, 11416–11427.

