 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yadira Palacios | -- | 3139 | 2023-07-01 04:23:49 | | | |
| 2 | Peter Tang | Meta information modification | 3139 | 2023-07-03 04:02:28 | | | | |
| 3 | Yadira Palacios | + 36 word(s) | 3175 | 2023-07-03 23:34:33 | | | | |
| 4 | Yadira Palacios | Meta information modification | 3175 | 2023-07-03 23:35:41 | | | | |
| 5 | Peter Tang | + 1 word(s) | 3176 | 2023-07-04 09:53:31 | | |
Video Upload Options
Mycobacterium tuberculosis (Mtb) modulates diverse cell death pathways to escape the host immune responses and favor its dissemination, a complex process of interest in pathogenesis-related studies. The main virulence factors of Mtb that alter cell death pathways are classified according to their origin as either non-protein (for instance, lipomannan) or protein (such as the PE family and ESX secretion system). The 38 kDa lipoprotein, ESAT-6 (early antigen-secreted protein 6 kDa), and another secreted protein, tuberculosis necrotizing toxin (TNT), induces necroptosis, thereby allowing mycobacteria to survive inside the cell. The inhibition of pyroptosis by blocking inflammasome activation by Zmp1 and PknF is another pathway that aids the intracellular replication of Mtb. Autophagy inhibition is another mechanism that allows Mtb to escape the immune response. The enhanced intracellular survival (Eis) protein, other proteins, such as ESX-1, SecA2, SapM, PE6, and certain microRNAs, also facilitate Mtb host immune escape process.
1. Introduction
2. Mtb Virulence Factors: Who Are Involved in Disturbing Cell Death?
2.1. Non-Protein Virulence Factors
Major Non-Protein Virulence Factors Inside the Cell Wall
-
Phosphatidyl-myo-inositol mannosides (PIMs) are the most abundant glycolipids in the mycobacterial cell envelope and are precursors of lipomannan (LM) and lipoarabinomannan (LAM) [17]. The PIMs comprise variable numbers of mannose units and levels of acylation. Virulent species possess PIMs with five or six mannoses that bind to the mannose receptor (MR), contributing to macrophage uptake. A few of the mannoses also interact with the dendritic cell (DC)-specific intercellular adhesion molecule-3-grabbing non-integrin DC-SIGN from DC [2][18]. Acyl phosphatidyl-myo-inositol dimannoside (AcPIM2) is a part of the inner membrane in most mycobacterial species, and AcPIM6 is involved in maintaining cell envelope integrity [16];
-
LM is a multi-glycosylated lipid or polymannosylated PIMs, which is the basic structure of LAM. LM efficiently activates the innate immune response via a tetra-acylated form that activates macrophages through toll-like receptor-2 and 4 (TLR2 and TLR4, respectively). In contrast, its di-acylated form regulates and inhibits nitric oxide (NO) production and cytokine secretion in activated macrophages [19];
-
LAM is a glycolipoconjugate composed of LM bound to multiple arabinose residues. When LAM acquires extra and random formation of mannose-capped LM, it is called ManLAM. An addition of phosphoinositol-capped LM makes it known as PILAM, and uncapped or arabinofuranosyl-terminated LM makes it AraLAM. PIMs, LM, LAM, and ManLAM, are recognized by specific receptors, expressed on the cell surface of antigen-presenting cells, such as MR, DC-SIGN, and dendritic cell activating receptor (DCAR), facilitating Mtb uptake into host cells [20][21]. Indeed, the diversity in Mtb mannosylated cell walls alters its virulence, affecting pathogeny and host adaptation [21];
-
Phthiocerol dimycocerosates (PDIM) are lipids present on the outer leaflet of the outer membrane that may be secreted or shed from mycobacteria. PDIM promotes Mtb’s evasion of TLR-mediated pathogen detection, delaying the recruitment of immune cells and adaptive responses to the infection site [22]. In addition, PDIM has been suggested to be crucial for phagocytosis and in the rupture of phagosomes and mitochondrial membranes [22][23].
2.2. Protein Virulence Factors
3. Cell Death Mechanisms Activated by Mtb Virulence Factors: The Good and the Bad
3.1. Apoptosis
3.2. Necrosis
3.3. Pyroptosis
3.4. Necroptosis
3.5. Autophagy-Dependent Cell Death
3.6. Ferroptosis
4. Final Considerations
Mtb exploits its varied virulence factors to orchestrate the infection process, facilitating its growth, dissemination, and latency. Host cell death is a critical mechanism that determines the outcome of infection. It is one of the main processes manipulated by mycobacterium to favor itself, thereby having various consequences on the host cell. It has been proposed that apoptosis and pyroptosis, which are the cell death mechanisms helpful for Mtb, restrict intracellular bacterial growth and facilitate anti-Mtb immune responses. Meanwhile, necroptosis and ferroptosis benefit Mtb replication and transmission (Figure 1).
The activation and inhibition of host cell death are driven by Mtb through its virulence factors; in fact, one virulence factor can be critical to more than one cell death modality, allowing the persistence of Mtb in the host.
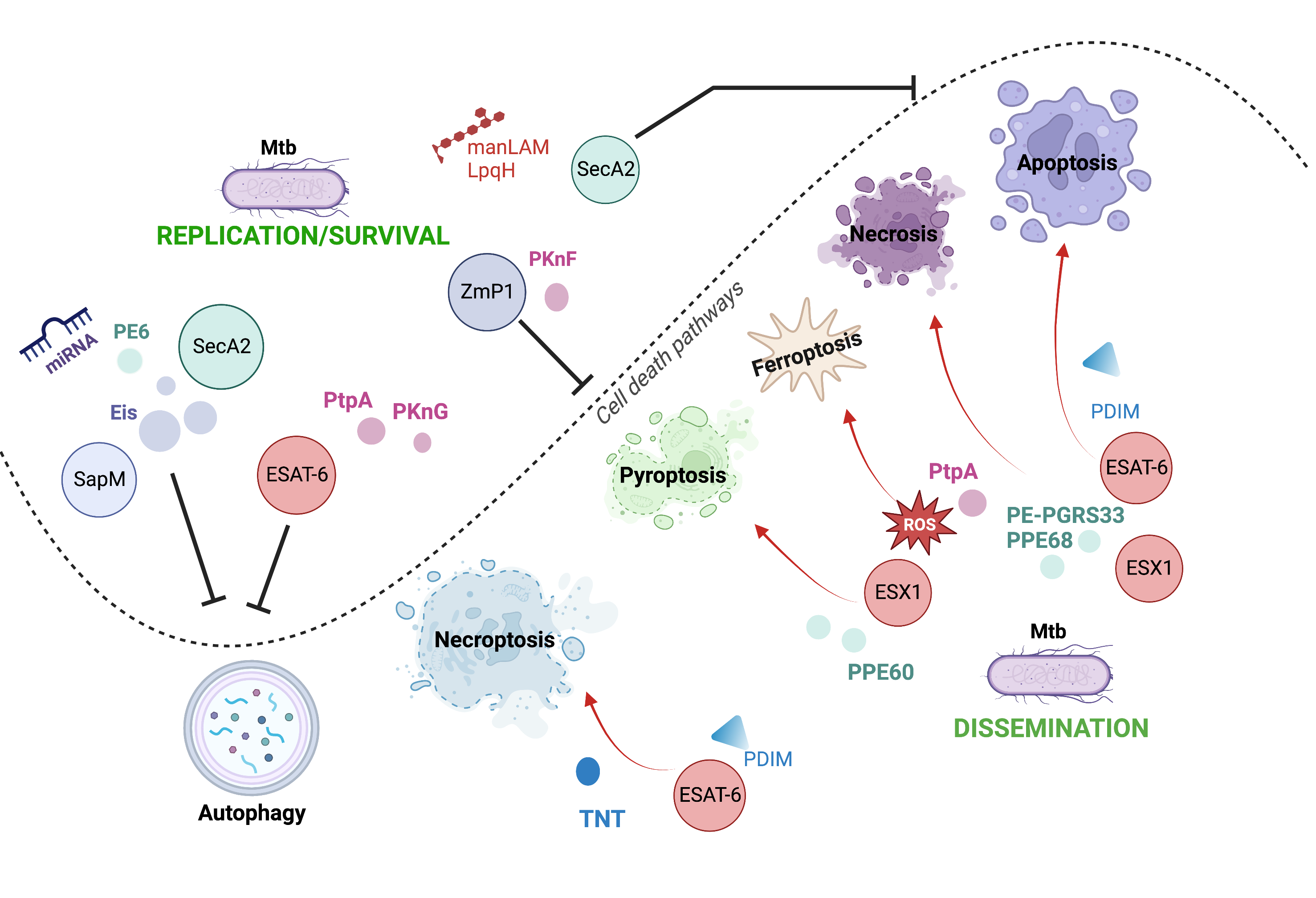
Figure 1. Cell death pathways modulated by Mtb through their virulence factors. Mtb can use its various virulence factors as inductors (red arrows) or inhibitors (black inhibitors) of diverse cell death mechanisms to orchestrate the infection process.
References
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Prim. 2016, 2, 16076.
- Echeverria-Valencia, G.; Flores-Villalva, S.; Espitia, C.I. Virulence Factors and Pathogenicity of Mycobacterium; Ribón, W., Ed.; IntechOpen: Rijeka, Croatia, 2017; p. Ch. 12. ISBN 978-1-78923-211-0.
- Global Tuberculosis Report 2022. Available online: https://www.who.int/publications/i/item/9789240061729 (accessed on 2 February 2023).
- Lee, J.; Boyce, S.; Powers, J.; Baer, C.; Sassetti, C.M.; Behar, S.M. CD11cHi Monocyte-Derived Macrophages Are a Major Cellular Compartment Infected by Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008621.
- Nadolinskaia, N.I.; Kotliarova, M.S.; Goncharenko, A.V. Fighting Tuberculosis: In Search of a BCG Replacement. Microorganisms 2022, 11, 51.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332.
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164.
- Mohareer, K.; Asalla, S.; Banerjee, S. Cell Death at the cross Roads of Host-Pathogen Interaction in Mycobacterium tuberculosis Infection. Tuberculosis 2018, 113, 99–121.
- Ramon-Luing, L.A.; Olvera, Y.; Flores-Gonzalez, J.; Palacios, Y.; Carranza, C.; Aguilar-Duran, Y.; Vargas, M.A.; Gutierrez, N.; Medina-Quero, K.; Chavez-Galan, L. Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis. Pathogens 2022, 11, 492.
- Feng, Z.; Bai, X.; Wang, T.; Garcia, C.; Bai, A.; Li, L.; Honda, J.R.; Nie, X.; Chan, E.D. Differential Responses by Human Macrophages to Infection With Mycobacterium tuberculosis and Non-Tuberculous Mycobacteria. Front. Microbiol. 2020, 11, 116.
- Butler, R.E.; Brodin, P.; Jang, J.; Jang, M.S.; Robertson, B.D.; Gicquel, B.; Stewart, G.R. The Balance of Apoptotic and Necrotic Cell Death in Mycobacterium tuberculosis Infected Macrophages Is Not Dependent on Bacterial Virulence. PLoS ONE 2012, 7, e47573.
- Coscolla, M.; Gagneux, S. Consequences of Genomic Diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444.
- Holzheimer, M.; Buter, J.; Minnaard, A.J. Chemical Synthesis of Cell Wall Constituents of Mycobacterium tuberculosis. Chem. Rev. 2021, 121, 9554–9643.
- Chiaradia, L.; Lefebvre, C.; Parra, J.; Marcoux, J.; Burlet-Schiltz, O.; Etienne, G.; Tropis, M.; Daffé, M. Dissecting the Mycobacterial Cell Envelope and Defining the Composition of the Native Mycomembrane. Sci. Rep. 2017, 7, 12807.
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and Virulence of Mycobacterium tuberculosis. Virulence 2023, 14, 2150449.
- Bansal-Mutalik, R.; Nikaido, H. Mycobacterial Outer Membrane Is a Lipid Bilayer and the Inner Membrane Is Unusually Rich in Diacyl Phosphatidylinositol Dimannosides. Proc. Natl. Acad. Sci. USA 2014, 111, 4958–4963.
- Torrelles, J.B.; Azad, A.K.; Schlesinger, L.S. Fine Discrimination in the Recognition of Individual Species of Phosphatidyl-Myo-Inositol Mannosides from Mycobacterium tuberculosis by C-Type Lectin Pattern Recognition Receptors. J. Immunol. 2006, 177, 1805–1816.
- Doz, E.; Rose, S.; Nigou, J.; Gilleron, M.; Puzo, G.; Erard, F.; Ryffel, B.; Quesniaux, V.F.J. Acylation Determines the Toll-like Receptor (TLR)-Dependent Positive versus TLR2-, Mannose Receptor-, and SIGNR1-Independent Negative Regulation of pro-Inflammatory Cytokines by Mycobacterial Lipomannan. J. Biol. Chem. 2007, 282, 26014–26025.
- Turner, J.; Torrelles, J.B. Mannose-Capped Lipoarabinomannan in Mycobacterium tuberculosis Pathogenesis. Pathog. Dis. 2018, 76, fty026.
- Torrelles, J.B.; Schlesinger, L.S. Diversity in Mycobacterium tuberculosis Mannosylated Cell Wall Determinants Impacts Adaptation to the Host. Tuberculosis 2010, 90, 84–93.
- Rens, C.; Chao, J.D.; Sexton, D.L.; Tocheva, E.I.; Av-Gay, Y. Roles for Phthiocerol Dimycocerosate Lipids in Mycobacterium tuberculosis Pathogenesis. Microbiology 2021, 167, 001042.
- Day, T.A.; Mittler, J.E.; Nixon, M.R.; Thompson, C.; Miner, M.D.; Hickey, M.J.; Liao, R.P.; Pang, J.M.; Shayakhmetov, D.M.; Sherman, D.R. Mycobacterium tuberculosis Strains Lacking Surface Lipid Phthiocerol Dimycocerosate Are Susceptible to Killing by an Early Innate Host Response. Infect. Immun. 2014, 82, 5214–5222.
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple Cell Death Modalities and Their Key Features (Review). World Acad. Sci. J. 2020, 2, 39–48.
- Grover, S.; Sharma, T.; Singh, Y.; Kohli, S.; Manjunath, P.; Singh, A.; Wieler, L.H.; Tedin, K.; Ehtesham, N.Z.; Hasnain, S.E.; et al. The PGRS Domain of Mycobacterium tuberculosis PE_PGRS Protein Rv0297 Is Involved in Endoplasmic Reticulum Stress-Mediated Apoptosis through Toll-like Receptor 4. mBio 2018, 9, e01017-18.
- Chai, Q.; Wang, L.; Liu, C.H.; Ge, B. New Insights into the Evasion of Host Innate Immunity by Mycobacterium tuberculosis. Cell. Mol. Immunol. 2020, 17, 901–913.
- Lossi, L. The Concept of Intrinsic versus Extrinsic Apoptosis. Biochem. J. 2022, 479, 357–384.
- Negulescu, A.M.; Mehlen, P. Dependence Receptors—The Dark Side Awakens. FEBS J. 2018, 285, 3909–3924.
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, Function, and Biotechnological Aspects. Biotechnol. Appl. Biochem. 2022, 69, 1633–1645.
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592.
- Li, X.; Li, F.; Zhang, X.; Zhang, H.; Zhao, Q.; Li, M.; Wu, X.; Wang, L.; Liu, J.; Wu, X.; et al. Caspase-8 Auto-Cleavage Regulates Programmed Cell Death and Collaborates with RIPK3/MLKL to Prevent Lymphopenia. Cell Death Differ. 2022, 29, 1500–1512.
- Li, Y.; Zhou, M.; Hu, Q.; Bai, X.C.; Huang, W.; Scheres, S.H.W.; Shi, Y. Mechanistic Insights into Caspase-9 Activation by the Structure of the Apoptosome Holoenzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1542–1547.
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100.
- Flores-Romero, H.; Hohorst, L.; John, M.; Albert, M.; King, L.E.; Beckmann, L.; Szabo, T.; Hertlein, V.; Luo, X.; Villunger, A.; et al. BCL-2-Family Protein TBID Can Act as a BAX-like Effector of Apoptosis. EMBO J. 2022, 41, e108690.
- Philips, J.A.; Ernst, J.D. Tuberculosis Pathogenesis and Immunity. Aging 2012, 7, 353–384.
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667.
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb Perspect. Biol. 2015, 7, a006080.
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by Caspase-3 during Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat. Commun. 2017, 8, 14128.
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma Membrane Changes during Programmed Cell Deaths. Cell Res. 2018, 28, 9–21.
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595.
- Rastogi, S.; Briken, V. Interaction of Mycobacteria with Host Cell Inflammasomes. Front. Immunol. 2022, 13, 791136.
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The Pyroptosome: A Supramolecular Assembly of ASC Dimers Mediating Inflammatory Cell Death via Caspase-1 Activation. Cell Death Differ. 2007, 14, 1590–1604.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665.
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673.
- Zhang, G.; Wang, J.; Zhao, Z.; Xin, T.; Fan, X.; Shen, Q.; Raheem, A.; Lee, C.R.; Jiang, H.; Ding, J. Regulated Necrosis, a Proinflammatory Cell Death, Potentially Counteracts Pathogenic Infections. Cell Death Dis. 2022, 13, 637.
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in Development and Diseases. Genes Dev. 2018, 32, 327–340.
- Chirieleison, S.M.; Kertesy, S.B.; Abbott, D.W. Synthetic Biology Reveals the Uniqueness of the RIP Kinase Domain. J. Immunol. 2016, 196, 4291.
- Ju, E.; Park, K.A.; Shen, H.M.; Hur, G.M. The Resurrection of RIP Kinase 1 as an Early Cell Death Checkpoint Regulator-a Potential Target for Therapy in the Necroptosis Era. Exp. Mol. Med. 2022, 54, 1401–1411.
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 Activation by TAK1-Mediated Phosphorylation Dictates Apoptosis and Necroptosis. Nat. Commun. 2017, 8, 359.
- Zhang, J.; Song, L.; Jia, J.; Tian, W.; Lai, R.; Zhang, Z.; Li, J.; Ju, J.; Xu, H. Knowledge Mapping of Necroptosis From 2012 to 2021: A Bibliometric Analysis. Front. Immunol. 2022, 13, 2930.
- Stutz, M.D.; Ojaimi, S.; Allison, C.; Preston, S.; Arandjelovic, P.; Hildebrand, J.M.; Sandow, J.J.; Webb, A.I.; Silke, J.; Alexander, W.S.; et al. Necroptotic Signaling Is Primed in Mycobacterium tuberculosis-Infected Macrophages, but Its Pathophysiological Consequence in Disease Is Restricted. Cell Death Differ. 2018, 25, 951–965.
- Zhang, L.; He, Y.; Jiang, Y.; Wu, Q.; Liu, Y.; Xie, Q.; Zou, Y.; Wu, J.; Zhang, C.; Zhou, Z.; et al. PRMT1 Reverts the Immune Escape of Necroptotic Colon Cancer through RIP3 Methylation. Cell Death Dis. 2023, 14, 233.
- Chen, D.; Tong, J.; Yang, L.; Wei, L.; Stolz, D.B.; Yu, J.; Zhang, J.; Zhang, L. PUMA Amplifies Necroptosis Signaling by Activating Cytosolic DNA Sensors. Proc. Natl. Acad. Sci. USA 2018, 115, 3930–3935.
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL Forms Cation Channels. Cell Res. 2016, 26, 517.
- Cai, Z.; Zhang, A.; Choksi, S.; Li, W.; Li, T.; Zhang, X.M.; Liu, Z.G. Activation of Cell-Surface Proteases Promotes Necroptosis, Inflammation and Cell Migration. Cell Res. 2016, 26, 886–900.
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed Necrotic Cell Death of Macrophages: Focus on Pyroptosis, Necroptosis, and Parthanatos. Redox Biol. 2019, 26, 101239.
- Seo, J.; Lee, E.W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.K.; Kim, J.H.; Han, S.Y.; et al. CHIP Controls Necroptosis through Ubiquitylation- and Lysosome-Dependent Degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302.
- Molnár, T.; Pallagi, P.; Tél, B.; Király, R.; Csoma, E.; Jenei, V.; Varga, Z.; Gogolák, P.; Odile Hueber, A.; Máté, Z.; et al. Caspase-9 Acts as a Regulator of Necroptotic Cell Death. FEBS J. 2021, 288, 6476–6491.
- Cao, W.; Li, J.; Yang, K.; Cao, D. An Overview of Autophagy: Mechanism, Regulation and Research Progress. Bull Cancer 2021, 108, 304–322.
- Deretic, V. Autophagy in Inflammation, Infection, and Immunometabolism. Immunity 2021, 54, 437–453.
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381.
- Nisa, A.; Kipper, F.C.; Panigrahy, D.; Tiwari, S.; Kupz, A.; Subbian, S. Different Modalities of Host Cell Death and Their Impact on Mycobacterium tuberculosis Infection. Am. J. Physiol.-Cell Physiol. 2022, 323, C1444–C1474.
- Bah, A.; Sanicas, M.; Nigou, J.; Guilhot, C.; Astarie-Dequeker, C.; Vergne, I. The Lipid Virulence Factors of Mycobacterium tuberculosis Exert Multilayered Control over Autophagy-Related Pathways in Infected Human Macrophages. Cells 2020, 9, 666.
- Lee, Y.J.; Kim, J.K.; Jung, C.H.; Kim, Y.J.; Jung, E.J.; Lee, S.H.; Choi, H.R.; Son, Y.S.; Shim, S.M.; Jeon, S.M.; et al. Chemical Modulation of SQSTM1/P62-Mediated Xenophagy That Targets a Broad Range of Pathogenic Bacteria. Autophagy 2022, 18, 2926–2945.
- Amaral, E.P.; Foreman, T.W.; Namasivayam, S.; Hilligan, K.L.; Kauffman, K.D.; Bomfim, C.C.B.; Costa, D.L.; Barreto-Duarte, B.; Gurgel-Rocha, C.; Santana, M.F.; et al. GPX4 Regulates Cellular Necrosis and Host Resistance in Mycobacterium tuberculosis Infection. J. Exp. Med. 2022, 219, e20220504.
- Yang, S.; Ouyang, J.; Lu, Y.; Harypursat, V.; Chen, Y. A Dual Role of Heme Oxygenase-1 in Tuberculosis. Front. Immunol. 2022, 13, 842858.



