Mycobacterium tuberculosis (Mtb) modulates diverse cell death pathways to escape the host immune responses and favor its dissemination, a complex process of interest in pathogenesis-related studies. The main virulence factors of Mtb that alter cell death pathways are classified according to their origin as either non-protein (for instance, lipomannan) or protein (such as the PE family and ESX secretion system). The 38 kDa lipoprotein, ESAT-6 (early antigen-secreted protein 6 kDa), and another secreted protein, tuberculosis necrotizing toxin (TNT), induces necroptosis, thereby allowing mycobacteria to survive inside the cell. The inhibition of pyroptosis by blocking inflammasome activation by Zmp1 and PknF is another pathway that aids the intracellular replication of Mtb. Autophagy inhibition is another mechanism that allows Mtb to escape the immune response. The enhanced intracellular survival (Eis) protein, other proteins, such as ESX-1, SecA2, SapM, PE6, and certain microRNAs, also facilitate Mtb host immune escape process.
- tuberculosis
- cell death
- virulence factors
- apoptosis
- necrosis
- pyroptosis
- necroptosis
1. Introduction
2. Mtb Virulence Factors: Who Are Involved in Disturbing Cell Death?
Virulence is the ability of a pathogen to cause disease [2]. To define Mtb virulence, the following factors should be considered: (1) the ability of a bacterium to avoid the host’s immune response; (2) its capacity to cause lung damage; and (3) its successful transmission to infect a new host [2][13]. Unlike other pathogens, such as Vibrio cholerae or Corynebacterium diphtheriae, Mtb does not use toxins and enzymes, which are the typical virulence factors; instead, several virulence-associated genes compensate for them [2]. Mycobacterial virulence factors can be divided based on their nature into (a) non-protein molecules, such as lipids, sugars, and (b) proteins. The following Mtb molecules can modify cell death pathways:2.1. Non-Protein Virulence Factors
Lipids, glycolipids, glycans, nucleic acids, and metabolites are included in this group; many are vital cell surface components involved in host–pathogen interaction, recognition, intracellular survival, and virulence [14].Major Non-Protein Virulence Factors Inside the Cell Wall
Mtb has a lipid-rich envelope essential for its survival and virulence [14]. The mycobacterial cell wall contains up to 60% lipids [15], and many cell wall components that are secreted, shed, or localized on the bacterial surface, interact with the host cells [16]. Before describing how virulence factors modulate the cell death pathway, a brief description of the more abundant non-protein ones is provided below:-
Phosphatidyl-myo-inositol mannosides (PIMs) are the most abundant glycolipids in the mycobacterial cell envelope and are precursors of lipomannan (LM) and lipoarabinomannan (LAM) [17]. The PIMs comprise variable numbers of mannose units and levels of acylation. Virulent species possess PIMs with five or six mannoses that bind to the mannose receptor (MR), contributing to macrophage uptake. A few of the mannoses also interact with the dendritic cell (DC)-specific intercellular adhesion molecule-3-grabbing non-integrin DC-SIGN from DC [2][18]. Acyl phosphatidyl-myo-inositol dimannoside (AcPIM2) is a part of the inner membrane in most mycobacterial species, and AcPIM6 is involved in maintaining cell envelope integrity [16];
-
LM is a multi-glycosylated lipid or polymannosylated PIMs, which is the basic structure of LAM. LM efficiently activates the innate immune response via a tetra-acylated form that activates macrophages through toll-like receptor-2 and 4 (TLR2 and TLR4, respectively). In contrast, its di-acylated form regulates and inhibits nitric oxide (NO) production and cytokine secretion in activated macrophages [19];
-
LAM is a glycolipoconjugate composed of LM bound to multiple arabinose residues. When LAM acquires extra and random formation of mannose-capped LM, it is called ManLAM. An addition of phosphoinositol-capped LM makes it known as PILAM, and uncapped or arabinofuranosyl-terminated LM makes it AraLAM. PIMs, LM, LAM, and ManLAM, are recognized by specific receptors, expressed on the cell surface of antigen-presenting cells, such as MR, DC-SIGN, and dendritic cell activating receptor (DCAR), facilitating Mtb uptake into host cells [20][21]. Indeed, the diversity in Mtb mannosylated cell walls alters its virulence, affecting pathogeny and host adaptation [21];
-
Phthiocerol dimycocerosates (PDIM) are lipids present on the outer leaflet of the outer membrane that may be secreted or shed from mycobacteria. PDIM promotes Mtb’s evasion of TLR-mediated pathogen detection, delaying the recruitment of immune cells and adaptive responses to the infection site [22]. In addition, PDIM has been suggested to be crucial for phagocytosis and in the rupture of phagosomes and mitochondrial membranes [22][23].
2.2. Protein Virulence Factors
Along with abundant lipids, the Mtb cell wall also contains proteins that directly affect the host’s immune response. Various reports have shown that protein families, such as PE, PPE, and lipoproteins, alter the host immune response. The effects include modulating cytokine production and arresting phagosome maturation, phagosome escape, autophagy, and cell death [2][16].3. Cell Death Mechanisms Activated by Mtb Virulence Factors: The Good and the Bad
Cell death was initially classified into three main types based on morphological, biochemical, and immunological criteria: type I (apoptosis); type II (autophagy); and type III (necrosis) [24]. Currently, novel types of cell death are characterized by a more integrative profile, including trigger stimuli, molecular mechanisms, and morphological, enzymological, and immunological characteristics. In 2018, the Nomenclature Committee on Cell Death (NCCD) proposed a new classification based on the mechanistic aspects of regulated cell death (RCD) and a definition based on the molecular and essential aspects. Some of these cell death types include intrinsic apoptosis, extrinsic apoptosis, mitochondrial permeability transition (MPT)-driven necrosis, necroptosis, ferroptosis, pyroptosis, autophagy-dependent cell death, parthanatos, endothelial cell death, NETotic cell death, lysosome-dependent cell death, immunogenic cell death, cellular senescence, and mitotic catastrophe [6]. Intriguingly, the molecular mechanisms involved in each RCD type have a considerable degree of interconnectivity, sharing key mediators. The novel signaling pathways involved in RCD are still being characterized. Apoptosis and necrosis can be considered as light and shadows in the immune response. Apoptosis is a “double-edged sword“ for the host because Mtb can inhibit or activate apoptosis for its benefit. Grover et al. showed that apoptosis disseminates Mtb infection in lung granulomas during later stages [25]. Necrosis enhances bacterial replication and facilitates reinfection [26]. Recently, it was reported that Mtb virulence factors block molecules of the BCL-2 family to inhibit conventional apoptosis. Moreover, it also simultaneously alters the levels of molecules involved in inducing apoptosis (related to RE stress), necroptosis, and pyroptosis; thus, this phenomenon might possibly be a strategy for Mtb to survive [10].3.1. Apoptosis
Apoptosis is a highly regulated cell death process, wherein a broad definition of molecular mechanisms allows it to be classified as type I cell death. It is characterized by cytoplasmic contraction, confinement of cytoplasmic contents, chromatin condensation (pyknosis), nuclear fragmentation (karyorrhexis), and blebs in the plasma membrane, forming tiny, apparently intact vesicles (known as apoptotic bodies) that can be removed by phagocytosis [6]. Furthermore, the nature of the stimuli may trigger two different apoptotic pathways, the extrinsic and intrinsic (or mitochondrial) pathways, with convergence, overlap, and synergism in different steps, mainly in the effector caspases [6]. The factors initiating the intrinsic pathway include cytotoxic stresses, such as radiation, toxins, hypoxia, reactive oxygen species, virus infection, and cytokine deprivation [25]. Extracellular alterations induce the extrinsic pathway, where cell-surface death receptors, such as FAS (also CD95 or APO-1), members of the TNF receptor superfamily [6][27], and dependence receptors (such as netrin receptors DCC and UNC5B) trigger apoptosis in the absence of ligands [28]. Extrinsic apoptosis mediated by the oligomerization of death receptor signaling may occur in two ways: (a) caspase-8/-10 activation, leading to caspase-3 activation to cleave critical proteins for cell survival and DNA fragmentation [29]; and (b) caspase-8 activation leading to BID cleavage and mitochondrial outer membrane permeabilization (MOMP) induction. It has also been suggested that extrinsic apoptosis mediated by dependent receptors leads to the activation of caspase-9 and caspase-3 [28]. After ligand-death receptor interactions, monomeric caspase-8 is recruited through its death-effector domain (DED) to the death-inducing signal complex (DISC) in the cytoplasmic domain of the death receptor [30]. In addition, a conformational change allows the association of the adapter protein FAS-associated death domain (FADD) or TNF receptor-associated death domain (TRADD), leading to caspase-8 dimerization and activation of effector caspases, thereby impacting the methods previously described to induce cell death [6][30]. Caspase-8 autocleavage is essential for apoptosis; however, this process also inhibits necroptosis and affects lymphocyte homeostasis via the RIPK3/ mixed lineage kinase domain-like protein (MLKL) signaling axis [31]. In the intrinsic apoptosis, the BH3-only proteins activate the Bcl-2 family members BAX and BAK, leading to MOMP, impact on loss of mitochondrial transmembrane potential (ΔΨm), and induce release to cytosol apoptotic mitochondrial factors, such as cytochrome c, which interacts with Apaf-1 (Apoptotic protease activating factor-1) via their WD domain. This interaction induces conformational changes to induce APAF-1 oligomerization and interaction with dATP. Apaf-1-CARD domains are exposed in the apoptosome complex to recruit and activate initiator caspase-9. The assembly of CARD domains occurs in two kinds of complexes depending on apoptosome localization, which stimulates caspase-9 catalytic activity and suppresses inhibitory CARD activity in free caspase-9 [27][32]. Some pro-apoptotic proteins, such as SMAC and OMI, may be released by MOMP, inhibiting the caspase inhibitor XIAP (X-linked inhibitor of apoptosis protein). Generating tBID (a BH3-only protein) by caspase-8 may link the intrinsic and extrinsic apoptotic pathways. However, tBID may promote MOMP, and proapoptotic factors are released independently of BAX and BAK but positively impact SMAC [33][34].3.2. Necrosis
Necrosis is considered to be a form of accidental cell death induced by pathological or physiological conditions, such as heat shock, mechanical stress, oxidative stress, inhibition of caspase activity, reduced levels of ATP, and radiation. This process encompasses several cell death modalities, commonly including the loss of plasma membrane integrity and the release of intracellular components that favor the inflammatory response, unlike that in apoptosis [7][35]. NCCD includes MPT-driven necrosis, a form of RCD induced by specific perturbations of the intracellular microenvironment triggered by severe oxidative stress and cytosolic Ca2+ overload [6]. An abrupt loss of the inner mitochondrial membrane’s impermeability to small solutes leads to a rapid Δψm dissipation and osmotic breakdown of both mitochondrial membranes and, consequently, RCD [36]. The presence of a necrotic morphology (i.e., rupture of the plasma membrane) can also be observed in the late stages of apoptotic or autophagic cell death by the absence of phagocytosis; apoptotic bodies may lose their integrity, and this is denominated as secondary necrosis caused by apoptosis or autophagy [37]. Secondary necrosis is not accidental but is controlled by a specific biochemical pathway regulated by caspase-3 and mediated by the GSDMD-related protein DFNA5 (deafness-associated tumor suppressor (GSDMD-related protein). Thus, this is a form of programmed necrosis similar to pyroptosis; caspase-3 cleaves DFNA5 to produce a necrosis-promoting DFNA5-N fragment that forms a large pore in the plasma membrane to release inflammatory molecules [38]. Other forms of regulated necrosis, including necroptosis, pyroptosis, and ferroptosis, have also been described.3.3. Pyroptosis
Pyroptosis is a type of RCD that depends on the formation of pores in the plasma membrane by members of the gasdermin protein family and is often induced by the inflammatory activation of caspases [6]. Thus, this inflammatory form of programmed necrosis occurs mainly in myeloid cells after activating the pattern recognition receptor (PRR) [39]. In humans, it is triggered by caspase-1 and/or caspase-4/-5, inflammasome activation, and the maturation and release of I-β and IL-18 [40][41]. The inflammasome is a multiprotein complex containing PRRs, such as TLRs, nucleotide-binding domains, and leucine-rich repeat-containing receptors (NLRs), and is Absent in Melanoma (AIM)-like receptors (ALRs). It is activated by pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) when host cells are exposed to microbial infections, stress, or tissue damage [41]. NLRP1, NLRP3, AIM2, and pyrin require an adaptor protein for inflammasome assembly, an apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), which facilitates the recruitment of pro-caspase-1 to the inflammasome complex [42]. NLRP3 is critical in bacterial, fungal, and viral infections and contributes to the host immune response [43]. The canonical activation of NRLP3 could be induced by two pathways: 1) by the detection of PAMPs, leading to up-regulated transcription of NLRP3, ASC, pro-IL-1β, pro-IL-18, and pro-caspase-11 by activation of NF-κB; or 2) by detection of DAMPS, such as abnormal levels of potassium (K+) efflux, chloride (Cl−) efflux, calcium (Ca2+) influx, oxidized mitochondrial DNA (ox-mtDNA), lysosomal rupture and intracellular ROS production. After recognizing PAMP or DAMP, the NLRP3 complex is oligomerized and assembled with ASC to recruit pro-caspase-1 for cleavage into active caspase-1. Finally, caspase-1 activates pro-IL-1β and pro-IL-18 via proteolytic cleavage into their active forms, IL-1β and IL-18, respectively [41]. As a result of inflammasome activation, caspase-1 cleaves Gasdermin D (GSDMD) into its pore-forming N-terminal fragment, GSDMD-N. Indeed, GSDMD is the primary executor of pyroptosis, forming pores in the plasma membrane resulting in pyroptosis [44][45]. Pyroptosis is another cell death mechanism that eliminates infected or compromised cells and plays a protective role in the host’s immune response [46].3.4. Necroptosis
Necroptosis is a highly regulated type of necrosis; this modality of RCD is triggered by perturbations of extracellular or intracellular homeostasis and is modulated by the kinase activity of the receptor-interacting protein 1 (RIPK1), RIPK3, and MLKL [6][47]. RIP Kinases play an essential role not only in inflammatory cell death even also in inflammatory signaling [48]. RIPK1 participates in TNF-induced NF-κB activation to regulate the switch between TNF-induced apoptosis and necroptosis, and when it is associated with RIPK3, necroptosis is induced [49][50]. Necroptosis can be triggered by impaired apoptosis, that is, the formation of necrosomes [51], by RIPK1-3; RIPK2 and RIPK3 are activated by TRADD, FAS, TNFR1, or PRRs. PPRs include TLR3, TLR4, and Z-DNA-binding protein 1 (ZBP1, also known as DAI) [49][52]. Additionally, RIPK3 can be activated following a domain-dependent interaction of the RIP-homotypic interaction motif (RHIM) due to the activation of TLR3 by double-stranded RNA (dsRNA) within endosomes and TLR4 activation by lipopolysaccharide (LPS) or DAMPs at the plasma membrane. RIPK3 catalyzes the phosphorylation of MLKL, leading to the formation of MLKL oligomers (trimers or tetramers) and their translocation to the plasma membrane, where they bind to specific phosphatidylinositol phosphate species [53][54]. MLKL promotes Ca2+ influx, probably by its target, the transient receptor potential cation channel, receptor subfamily M member 7 (TRPM7). When MLK is present on the membrane, the ADAM family proteases promote the shedding of membrane-associated proteins and form Mg2+ channels [55][56]. Some of the factors that regulate necrosome formation include stress-induced phosphoprotein 1 (STIP1) protein, homology, U-box containing protein 1 (STUB1 or CHIP), A20 protein, Mg2+/Mn2+-dependent protein phosphatase 1 B (PPM1B), aurora kinase A (AURKA), and RIPK3 [57][58]. STIP1 and STUB1/CHIP degrade RIPK1 and RIPK3 via ubiquitination and lysosomal degradation. A20 inhibits necrosome formation by eliminating RIPK3 ubiquitination. PPM1B prevents MLKL from binding to the necrosome by reducing RIPK3 phosphorylation. Finally, AURKA physically interacts with RIPK1 and RIPK3 to inhibit the necrosomal function [58][59]. These negative necrosome regulators play an essential role in regulating necroptosis and may modulate the pathogenesis of various diseases.3.5. Autophagy-Dependent Cell Death
Autophagy-dependent cell death is a form of RCD that mechanistically depends on the autophagic machinery [6], an evolutionarily conserved catabolic process of eukaryotes, to maintain homeostasis in response to different stress conditions, such as starvation, hypoxia, absence of growth factors, and infection [60]. During autophagy, double-membrane vesicles, called autophagosomes, fuse with acidic compartments, or lysosomes, to give rise to autolysosomes that remove unwanted components from the cell, such as long half-life proteins, damaged organelles, and intracellular pathogens, such as Mtb. This phenomenon allows the cells to economize on macromolecule synthesis and increases cell survival [60][61]. Three types of autophagy have been described: chaperone-mediated autophagy (CMA); microautophagy; and macroautophagy. During CMA, cytosolic chaperones interact with soluble proteins and bind to the lysosomal receptor LAMP-2A (lysosome-associated membrane protein type 2a) for transportation to the lysosome and degradation of proteins [60][62]. Microautophagy involves the direct immersion of the material to be degraded by the lysosome through the lysosomal membrane’s extension, protrusion, and septation [63]. Macroautophagy, also known as the classical autophagy pathway, sequesters cytoplasmic portions and is named according to the content or organelle to be eliminated; for instance, when it contains intracellular pathogens, it is called xenophagy; when it contains mitochondria, it is called mitophagy and lysophagy [61][64][65].3.6. Ferroptosis
Ferroptosis is another form of RCD triggered by oxidative perturbations of the intracellular microenvironment under the constitutive control of glutathione peroxidase 4 (GPX4). Severe lipid peroxidation mainly depends on ROS generation and iron availability. Iron chelators and lipophilic antioxidants can inhibit ferroptosis [6]. Ferroptosis is regulated by molecules of the metabolic pathways that regulate cysteine exploitation, glutathione status, and nicotinamide adenine dinucleotide phosphate (NADP) function. Unlike other forms of RCD, such as apoptosis and necroptosis, ferroptosis is independent of caspases, necrosome components, and autophagy. Instead, ferroptotic cells display necrotic morphology with mitochondrial alterations, shrinkage, and electrodense ultrastructure. Additionally, they may release damage-associated molecular patterns (DAMPs) [66][67].4. Final Considerations
Mtb exploits its varied virulence factors to orchestrate the infection process, facilitating its growth, dissemination, and latency. Host cell death is a critical mechanism that determines the outcome of infection. It is one of the main processes manipulated by mycobacterium to favor itself, thereby having various consequences on the host cell. It has been proposed that apoptosis and pyroptosis, which are the cell death mechanisms helpful for Mtb, restrict intracellular bacterial growth and facilitate anti-Mtb immune responses. Meanwhile, necroptosis and ferroptosis benefit Mtb replication and transmission (Figure 1).
The activation and inhibition of host cell death are driven by Mtb through its virulence factors; in fact, one virulence factor can be critical to more than one cell death modality, allowing the persistence of Mtb in the host.
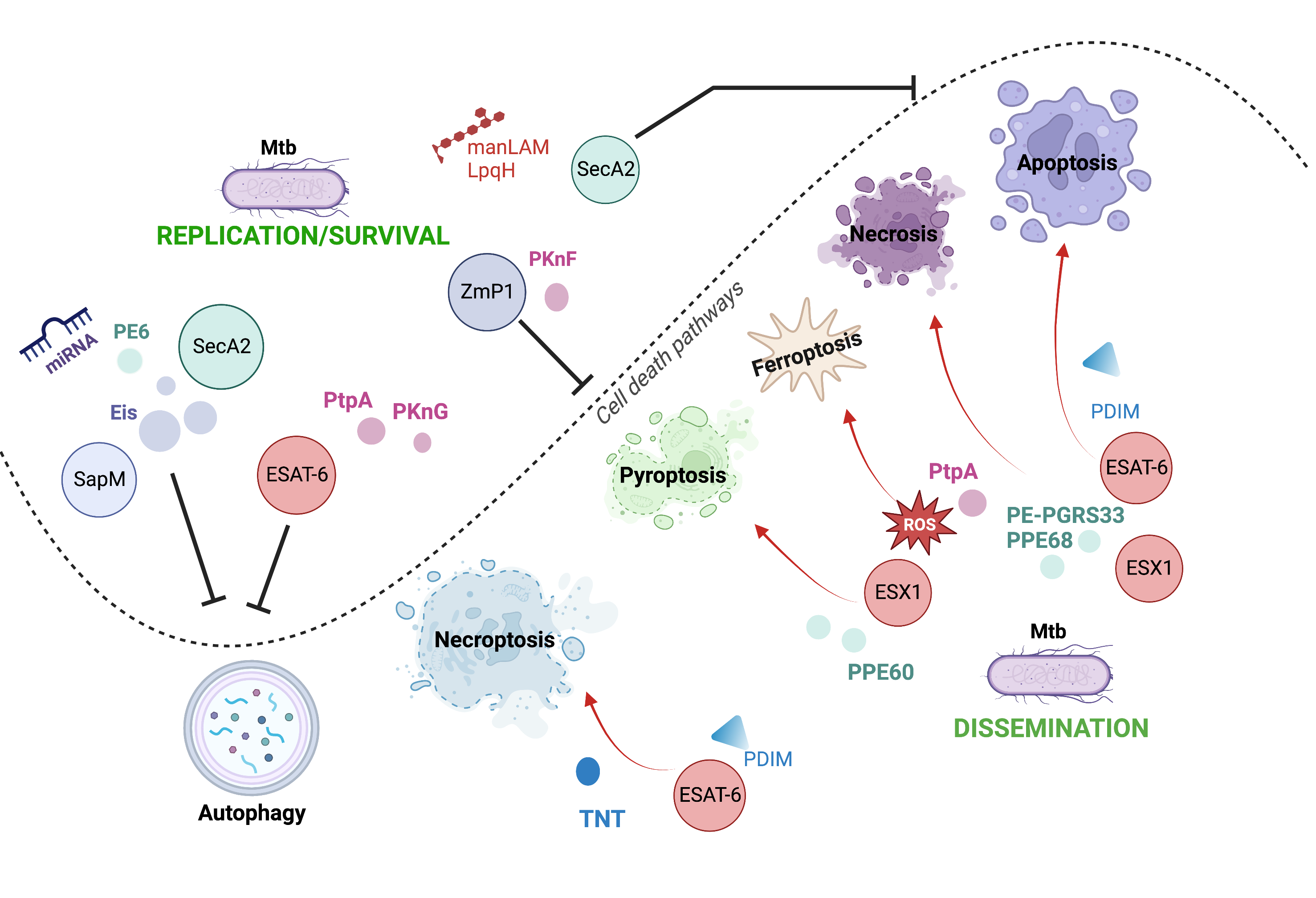
Figure 1. Cell death pathways modulated by Mtb through their virulence factors. Mtb can use its various virulence factors as inductors (red arrows) or inhibitors (black inhibitors) of diverse cell death mechanisms to orchestrate the infection process.
References
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Prim. 2016, 2, 16076.
- Echeverria-Valencia, G.; Flores-Villalva, S.; Espitia, C.I. Virulence Factors and Pathogenicity of Mycobacterium; Ribón, W., Ed.; IntechOpen: Rijeka, Croatia, 2017; p. Ch. 12. ISBN 978-1-78923-211-0.
- Global Tuberculosis Report 2022. Available online: https://www.who.int/publications/i/item/9789240061729 (accessed on 2 February 2023).
- Lee, J.; Boyce, S.; Powers, J.; Baer, C.; Sassetti, C.M.; Behar, S.M. CD11cHi Monocyte-Derived Macrophages Are a Major Cellular Compartment Infected by Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008621.
- Nadolinskaia, N.I.; Kotliarova, M.S.; Goncharenko, A.V. Fighting Tuberculosis: In Search of a BCG Replacement. Microorganisms 2022, 11, 51.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332.
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164.
- Mohareer, K.; Asalla, S.; Banerjee, S. Cell Death at the cross Roads of Host-Pathogen Interaction in Mycobacterium tuberculosis Infection. Tuberculosis 2018, 113, 99–121.
- Ramon-Luing, L.A.; Olvera, Y.; Flores-Gonzalez, J.; Palacios, Y.; Carranza, C.; Aguilar-Duran, Y.; Vargas, M.A.; Gutierrez, N.; Medina-Quero, K.; Chavez-Galan, L. Diverse Cell Death Mechanisms Are Simultaneously Activated in Macrophages Infected by Virulent Mycobacterium tuberculosis. Pathogens 2022, 11, 492.
- Feng, Z.; Bai, X.; Wang, T.; Garcia, C.; Bai, A.; Li, L.; Honda, J.R.; Nie, X.; Chan, E.D. Differential Responses by Human Macrophages to Infection With Mycobacterium tuberculosis and Non-Tuberculous Mycobacteria. Front. Microbiol. 2020, 11, 116.
- Butler, R.E.; Brodin, P.; Jang, J.; Jang, M.S.; Robertson, B.D.; Gicquel, B.; Stewart, G.R. The Balance of Apoptotic and Necrotic Cell Death in Mycobacterium tuberculosis Infected Macrophages Is Not Dependent on Bacterial Virulence. PLoS ONE 2012, 7, e47573.
- Coscolla, M.; Gagneux, S. Consequences of Genomic Diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444.
- Holzheimer, M.; Buter, J.; Minnaard, A.J. Chemical Synthesis of Cell Wall Constituents of Mycobacterium tuberculosis. Chem. Rev. 2021, 121, 9554–9643.
- Chiaradia, L.; Lefebvre, C.; Parra, J.; Marcoux, J.; Burlet-Schiltz, O.; Etienne, G.; Tropis, M.; Daffé, M. Dissecting the Mycobacterial Cell Envelope and Defining the Composition of the Native Mycomembrane. Sci. Rep. 2017, 7, 12807.
- Rahlwes, K.C.; Dias, B.R.S.; Campos, P.C.; Alvarez-Arguedas, S.; Shiloh, M.U. Pathogenicity and Virulence of Mycobacterium tuberculosis. Virulence 2023, 14, 2150449.
- Bansal-Mutalik, R.; Nikaido, H. Mycobacterial Outer Membrane Is a Lipid Bilayer and the Inner Membrane Is Unusually Rich in Diacyl Phosphatidylinositol Dimannosides. Proc. Natl. Acad. Sci. USA 2014, 111, 4958–4963.
- Torrelles, J.B.; Azad, A.K.; Schlesinger, L.S. Fine Discrimination in the Recognition of Individual Species of Phosphatidyl-Myo-Inositol Mannosides from Mycobacterium tuberculosis by C-Type Lectin Pattern Recognition Receptors. J. Immunol. 2006, 177, 1805–1816.
- Doz, E.; Rose, S.; Nigou, J.; Gilleron, M.; Puzo, G.; Erard, F.; Ryffel, B.; Quesniaux, V.F.J. Acylation Determines the Toll-like Receptor (TLR)-Dependent Positive versus TLR2-, Mannose Receptor-, and SIGNR1-Independent Negative Regulation of pro-Inflammatory Cytokines by Mycobacterial Lipomannan. J. Biol. Chem. 2007, 282, 26014–26025.
- Turner, J.; Torrelles, J.B. Mannose-Capped Lipoarabinomannan in Mycobacterium tuberculosis Pathogenesis. Pathog. Dis. 2018, 76, fty026.
- Torrelles, J.B.; Schlesinger, L.S. Diversity in Mycobacterium tuberculosis Mannosylated Cell Wall Determinants Impacts Adaptation to the Host. Tuberculosis 2010, 90, 84–93.
- Rens, C.; Chao, J.D.; Sexton, D.L.; Tocheva, E.I.; Av-Gay, Y. Roles for Phthiocerol Dimycocerosate Lipids in Mycobacterium tuberculosis Pathogenesis. Microbiology 2021, 167, 001042.
- Day, T.A.; Mittler, J.E.; Nixon, M.R.; Thompson, C.; Miner, M.D.; Hickey, M.J.; Liao, R.P.; Pang, J.M.; Shayakhmetov, D.M.; Sherman, D.R. Mycobacterium tuberculosis Strains Lacking Surface Lipid Phthiocerol Dimycocerosate Are Susceptible to Killing by an Early Innate Host Response. Infect. Immun. 2014, 82, 5214–5222.
- Yan, G.; Elbadawi, M.; Efferth, T. Multiple Cell Death Modalities and Their Key Features (Review). World Acad. Sci. J. 2020, 2, 39–48.
- Grover, S.; Sharma, T.; Singh, Y.; Kohli, S.; Manjunath, P.; Singh, A.; Wieler, L.H.; Tedin, K.; Ehtesham, N.Z.; Hasnain, S.E.; et al. The PGRS Domain of Mycobacterium tuberculosis PE_PGRS Protein Rv0297 Is Involved in Endoplasmic Reticulum Stress-Mediated Apoptosis through Toll-like Receptor 4. mBio 2018, 9, e01017-18.
- Chai, Q.; Wang, L.; Liu, C.H.; Ge, B. New Insights into the Evasion of Host Innate Immunity by Mycobacterium tuberculosis. Cell. Mol. Immunol. 2020, 17, 901–913.
- Lossi, L. The Concept of Intrinsic versus Extrinsic Apoptosis. Biochem. J. 2022, 479, 357–384.
- Negulescu, A.M.; Mehlen, P. Dependence Receptors—The Dark Side Awakens. FEBS J. 2018, 285, 3909–3924.
- Asadi, M.; Taghizadeh, S.; Kaviani, E.; Vakili, O.; Taheri-Anganeh, M.; Tahamtan, M.; Savardashtaki, A. Caspase-3: Structure, Function, and Biotechnological Aspects. Biotechnol. Appl. Biochem. 2022, 69, 1633–1645.
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592.
- Li, X.; Li, F.; Zhang, X.; Zhang, H.; Zhao, Q.; Li, M.; Wu, X.; Wang, L.; Liu, J.; Wu, X.; et al. Caspase-8 Auto-Cleavage Regulates Programmed Cell Death and Collaborates with RIPK3/MLKL to Prevent Lymphopenia. Cell Death Differ. 2022, 29, 1500–1512.
- Li, Y.; Zhou, M.; Hu, Q.; Bai, X.C.; Huang, W.; Scheres, S.H.W.; Shi, Y. Mechanistic Insights into Caspase-9 Activation by the Structure of the Apoptosome Holoenzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1542–1547.
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100.
- Flores-Romero, H.; Hohorst, L.; John, M.; Albert, M.; King, L.E.; Beckmann, L.; Szabo, T.; Hertlein, V.; Luo, X.; Villunger, A.; et al. BCL-2-Family Protein TBID Can Act as a BAX-like Effector of Apoptosis. EMBO J. 2022, 41, e108690.
- Philips, J.A.; Ernst, J.D. Tuberculosis Pathogenesis and Immunity. Aging 2012, 7, 353–384.
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667.
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb Perspect. Biol. 2015, 7, a006080.
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by Caspase-3 during Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat. Commun. 2017, 8, 14128.
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma Membrane Changes during Programmed Cell Deaths. Cell Res. 2018, 28, 9–21.
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595.
- Rastogi, S.; Briken, V. Interaction of Mycobacteria with Host Cell Inflammasomes. Front. Immunol. 2022, 13, 791136.
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The Pyroptosome: A Supramolecular Assembly of ASC Dimers Mediating Inflammatory Cell Death via Caspase-1 Activation. Cell Death Differ. 2007, 14, 1590–1604.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665.
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673.
- Zhang, G.; Wang, J.; Zhao, Z.; Xin, T.; Fan, X.; Shen, Q.; Raheem, A.; Lee, C.R.; Jiang, H.; Ding, J. Regulated Necrosis, a Proinflammatory Cell Death, Potentially Counteracts Pathogenic Infections. Cell Death Dis. 2022, 13, 637.
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in Development and Diseases. Genes Dev. 2018, 32, 327–340.
- Chirieleison, S.M.; Kertesy, S.B.; Abbott, D.W. Synthetic Biology Reveals the Uniqueness of the RIP Kinase Domain. J. Immunol. 2016, 196, 4291.
- Ju, E.; Park, K.A.; Shen, H.M.; Hur, G.M. The Resurrection of RIP Kinase 1 as an Early Cell Death Checkpoint Regulator-a Potential Target for Therapy in the Necroptosis Era. Exp. Mol. Med. 2022, 54, 1401–1411.
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 Activation by TAK1-Mediated Phosphorylation Dictates Apoptosis and Necroptosis. Nat. Commun. 2017, 8, 359.
- Zhang, J.; Song, L.; Jia, J.; Tian, W.; Lai, R.; Zhang, Z.; Li, J.; Ju, J.; Xu, H. Knowledge Mapping of Necroptosis From 2012 to 2021: A Bibliometric Analysis. Front. Immunol. 2022, 13, 2930.
- Stutz, M.D.; Ojaimi, S.; Allison, C.; Preston, S.; Arandjelovic, P.; Hildebrand, J.M.; Sandow, J.J.; Webb, A.I.; Silke, J.; Alexander, W.S.; et al. Necroptotic Signaling Is Primed in Mycobacterium tuberculosis-Infected Macrophages, but Its Pathophysiological Consequence in Disease Is Restricted. Cell Death Differ. 2018, 25, 951–965.
- Zhang, L.; He, Y.; Jiang, Y.; Wu, Q.; Liu, Y.; Xie, Q.; Zou, Y.; Wu, J.; Zhang, C.; Zhou, Z.; et al. PRMT1 Reverts the Immune Escape of Necroptotic Colon Cancer through RIP3 Methylation. Cell Death Dis. 2023, 14, 233.
- Chen, D.; Tong, J.; Yang, L.; Wei, L.; Stolz, D.B.; Yu, J.; Zhang, J.; Zhang, L. PUMA Amplifies Necroptosis Signaling by Activating Cytosolic DNA Sensors. Proc. Natl. Acad. Sci. USA 2018, 115, 3930–3935.
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL Forms Cation Channels. Cell Res. 2016, 26, 517.
- Cai, Z.; Zhang, A.; Choksi, S.; Li, W.; Li, T.; Zhang, X.M.; Liu, Z.G. Activation of Cell-Surface Proteases Promotes Necroptosis, Inflammation and Cell Migration. Cell Res. 2016, 26, 886–900.
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed Necrotic Cell Death of Macrophages: Focus on Pyroptosis, Necroptosis, and Parthanatos. Redox Biol. 2019, 26, 101239.
- Seo, J.; Lee, E.W.; Sung, H.; Seong, D.; Dondelinger, Y.; Shin, J.; Jeong, M.; Lee, H.K.; Kim, J.H.; Han, S.Y.; et al. CHIP Controls Necroptosis through Ubiquitylation- and Lysosome-Dependent Degradation of RIPK3. Nat. Cell Biol. 2016, 18, 291–302.
- Molnár, T.; Pallagi, P.; Tél, B.; Király, R.; Csoma, E.; Jenei, V.; Varga, Z.; Gogolák, P.; Odile Hueber, A.; Máté, Z.; et al. Caspase-9 Acts as a Regulator of Necroptotic Cell Death. FEBS J. 2021, 288, 6476–6491.
- Cao, W.; Li, J.; Yang, K.; Cao, D. An Overview of Autophagy: Mechanism, Regulation and Research Progress. Bull Cancer 2021, 108, 304–322.
- Deretic, V. Autophagy in Inflammation, Infection, and Immunometabolism. Immunity 2021, 54, 437–453.
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381.
- Nisa, A.; Kipper, F.C.; Panigrahy, D.; Tiwari, S.; Kupz, A.; Subbian, S. Different Modalities of Host Cell Death and Their Impact on Mycobacterium tuberculosis Infection. Am. J. Physiol.-Cell Physiol. 2022, 323, C1444–C1474.
- Bah, A.; Sanicas, M.; Nigou, J.; Guilhot, C.; Astarie-Dequeker, C.; Vergne, I. The Lipid Virulence Factors of Mycobacterium tuberculosis Exert Multilayered Control over Autophagy-Related Pathways in Infected Human Macrophages. Cells 2020, 9, 666.
- Lee, Y.J.; Kim, J.K.; Jung, C.H.; Kim, Y.J.; Jung, E.J.; Lee, S.H.; Choi, H.R.; Son, Y.S.; Shim, S.M.; Jeon, S.M.; et al. Chemical Modulation of SQSTM1/P62-Mediated Xenophagy That Targets a Broad Range of Pathogenic Bacteria. Autophagy 2022, 18, 2926–2945.
- Amaral, E.P.; Foreman, T.W.; Namasivayam, S.; Hilligan, K.L.; Kauffman, K.D.; Bomfim, C.C.B.; Costa, D.L.; Barreto-Duarte, B.; Gurgel-Rocha, C.; Santana, M.F.; et al. GPX4 Regulates Cellular Necrosis and Host Resistance in Mycobacterium tuberculosis Infection. J. Exp. Med. 2022, 219, e20220504.
- Yang, S.; Ouyang, J.; Lu, Y.; Harypursat, V.; Chen, Y. A Dual Role of Heme Oxygenase-1 in Tuberculosis. Front. Immunol. 2022, 13, 842858.