Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Edith Marianne Schneider Gasser | -- | 3111 | 2023-06-27 12:27:59 | | | |
| 2 | Markus Thiersch | Meta information modification | 3111 | 2023-06-28 11:09:31 | | | | |
| 3 | Fanny Huang | Meta information modification | 3111 | 2023-06-29 07:36:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Aboouf, M.A.; Thiersch, M.; Soliz, J.; Gassmann, M.; Schneider Gasser, E.M. The Brain at High Altitude. Encyclopedia. Available online: https://encyclopedia.pub/entry/46117 (accessed on 23 July 2026).
Aboouf MA, Thiersch M, Soliz J, Gassmann M, Schneider Gasser EM. The Brain at High Altitude. Encyclopedia. Available at: https://encyclopedia.pub/entry/46117. Accessed July 23, 2026.
Aboouf, Mostafa A., Markus Thiersch, Jorge Soliz, Max Gassmann, Edith M. Schneider Gasser. "The Brain at High Altitude" Encyclopedia, https://encyclopedia.pub/entry/46117 (accessed July 23, 2026).
Aboouf, M.A., Thiersch, M., Soliz, J., Gassmann, M., & Schneider Gasser, E.M. (2023, June 27). The Brain at High Altitude. In Encyclopedia. https://encyclopedia.pub/entry/46117
Aboouf, Mostafa A., et al. "The Brain at High Altitude." Encyclopedia. Web. 27 June, 2023.
Copy Citation
The brain requires over one-fifth of the total body oxygen demand for normal functioning. At high altitude (HA), the lower atmospheric oxygen pressure inevitably challenges the brain, affecting voluntary spatial attention, cognitive processing, and attention speed after short-term, long-term, or lifespan exposure. Molecular responses to HA are controlled mainly by hypoxia-inducible factors.
hypoxia
HIF
EPO
high altitude
1. High Altitude and Cognition
High altitude (HA) (defined as an altitude above 2500 m above sea level) is characterized by multiple harsh environmental conditions. Most physiological adaptations occur in response to the reduced atmospheric pressure, resulting in reduced oxygen partial pressure and causing reduced blood oxygenation saturation (SpO2) hypoxemia. The brain is susceptible to alterations in oxygen supply. Thus, HA exposure causes adverse changes in mood states, such as depression [1] and anxiety [2], and neurocognitive alterations, such as memory impairment [3] and attention disorders after both short- and long-term HA exposure [4][5]. Although numerous reports concern the physiological and neurological alteration that occurs after ascent to HA, less has been investigated on the cognitive and brain alterations in long-term and permanent inhabitants of HA.
Brain functions are affected by hypoxia not only after ascent to HA [6] but also after long-term exposure at a HA [7] and in native highlanders [8]. In unacclimatized individuals exposed to HA, sleep patterns can already be affected at elevations above 1600 m, changes in mood states like euphoria or depression are observed in some individuals from 2500 m on, and above 3000 m subjects can experience headache, dizziness, and confusion. Mood state alterations, including euphoria, quarrelsome, irritability, and apathy, occur temporarily after fast acute exposure to HA and return to baseline states after 48 to 52 h [9][10][11]. In contrast, short- and long-term exposure to HA causes biological, inflammatory, and structural brain changes that increase the risk of experiencing anxiety and depression symptoms [12] and neurocognitive dysfunctions such as slower reaction times, reduced attention (>3500 m), impaired learning, spatial and working memory (>4000 m), and impaired retrieval (>5500 m) (Figure 1) [7][8][13][14].
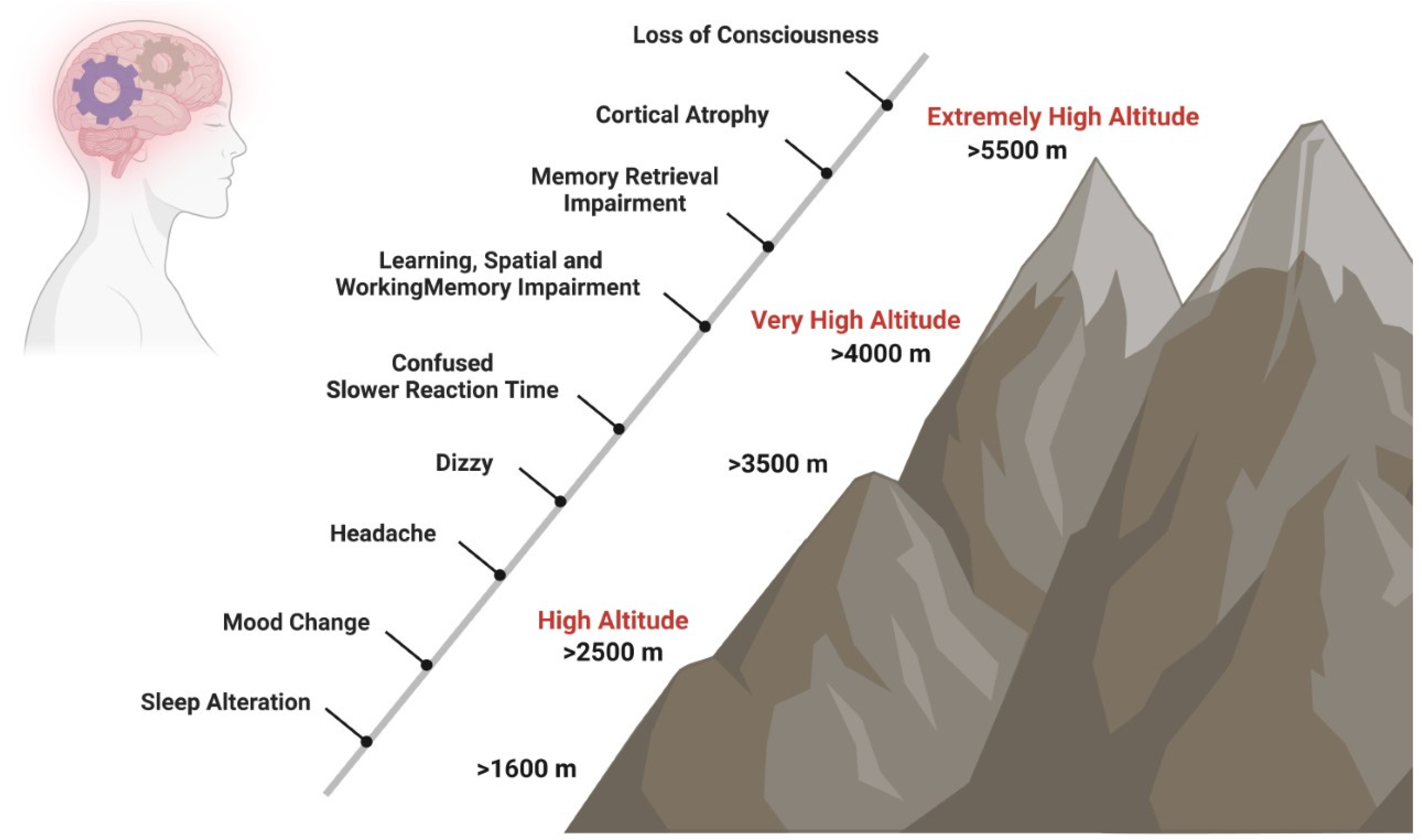
Figure 1. Impact of HA on cognition. Short- and long-term exposure to HA has effects on neurocognitive functions. Sleep patterns can be affected at elevations above 1600 m, changes in mood states like euphoria or depression are observed in some individuals from 2500 m on, and above 3000 m, subjects can experience headache, dizziness, and confusion. At very HA, slower reaction times, such as reduced attention (>3500 m), impaired learning, spatial and working memory (>4000 m), and impaired retrieval (>5500 m) may occur.
Mood alterations after HA exposure are attributed to changes in brain levels of dopamine and serotonin. Euphoria is attributed to increased brain dopamine levels [15], while lower serotonin levels are attributed to sadness, grief, worry, anxiety, and depression [16]. Studies in rodents and piglets have shown increased extracellular dopamine levels in the striatum when the oxygen pressure in the cerebral cortex decreases [17][18]. Moreover, reduced serotonin (5-hydroxytryptamine, 5-HT) availability is observed at HA in people with anxiety and depression, particularly in women [19]. Long-term exposure to HA induces a hypoxic stress response in which the noradrenergic brain regions, particularly the locus coeruleus and hypothalamic-pituitary-adrenal (HPA) axis, are involved [20]. Similarly, HA leads to alterations in brain bioenergetics, which could also contribute to depressive symptoms. Indeed, multiple magnetic resonance spectroscopy studies suggest that persons at HA with depression exhibit alteration in adenosine triphosphate (ATP) expression [21]. Similarly, an imbalance in mitochondrial dynamics has been demonstrated in the rat brain hippocampus after hypobaric hypoxia exposure [22]. Moreover, HA residents (1400 m, Salt Lake City in Utah, USA) exhibit significant differences in brain pH and inorganic phosphate levels compared with sea-level residents (13 m, Belmont City in Massachusetts, USA) [23].
Acute, subacute, and repeated HA exposure causes a reduction in neurocognitive processing speed (reaction time) and attention (switching, visual processing). A study in healthy subjects showed that acclimatization improves reaction time and attention due to increased SpO2. Thus, processing speed and attention are associated with SpO2 acclimatization [13].
Despite about 2.2% of the world’s population (more than 140 million people) living permanently at a HA [24], the impact of reduced SpO2 on cognition remains poorly explored. SpO2 values decrease with increasing altitude to a median of 96% (95–97) at 2500 m, 92% (90–93) at 3600 m, 87% (85–89) at 4100, and 81% (78–84) at 5100 m, with increasing variability at higher altitudes [25]. The reduced SpO2 at HAs induces a rapid increase in hypoxic ventilatory response (HVR) [26], but also breathing instability (periods of deep and rapid breathing) and central apnea during sleep [27]. Although the rate of sleep apnea is lower at HA than at sea level [28], the gap in SpO2 between wakefulness and sleep states is greater than at sea level [29][30]. The statistical distribution of SpO2 in children at HA shows that one out of four children saturates significantly less than the others [31]. Moreover, in one out of four children at HA, the oxygen desaturation index during sleep (which reflects SpO2 variability) is lower [32][33]. Changes in sleep patterns can modulate mood and cause deficits in working memory, attention, or executive functions [34]. Yet, whether low SpO2 during sleep correlates with impaired cognition at HA remains unknown.
Acute exposure to HA causes impairment in learning and memory encoding and retention [35]. Those alterations are also observed in lowlanders living at HA [36]. With the development of cognitive neuroscience research techniques such as functional magnetic resonance imaging (fMRI) and event-related potentials (ERP), it is possible now to explore the mechanisms underlying the effects of long-term HA exposure on cognition. After natural selection, highlanders have genetic and physiological adaptations to HA. Thus, brain alterations in HA-exposed lowlanders may not reflect the brain of adapted highlanders. Exposure to HA affects mainly visual spatial attention in lowlanders [37]. After the ascent to very HA, brain edema and cortical atrophy were reported in climbers [38], but the reduction in brain volume was only reported after more than three weeks of very HA exposure (>5500 m) [39]. For HA immigrants, an increase in gray matter is observed, probably associated with increased neurogenesis and vasculature. Alterations in white matter volumes after acute HA exposure were rarely observed, but diffuse imaging suggested injury [40]. Highlander descendants of Han immigrants showed increased interhemispheric fibers [41]. The changes in cerebrospinal fluid were very variable across studies [42].
fMRI is used to measure changes in blood dynamics caused by neuronal activity. Blood oxygenation level-dependent (BOLD) images and ERP studies showed a reduction in oxygenation and activity in the occipital visual cortex, insular cortex, cingulate cortex, precentral gyrus, hippocampus, and cerebellum in individuals staying at HA or HA natives [42].
Electroencephalogram (EEG) is used to evaluate the electrophysiological processing of cognitive activity, mostly after ascent to HA and in HA immigrants, but studies in HA natives are rare. In general, EEG shows that prolonged exposure to HA mainly impairs attention. In native adolescents in Bolivia, reductions in delta and beta frequency amplitude were recorded [43]. In Tibetan natives, a higher executive ability and impaired orienting ability were observed only at HA above 4000 m, suggesting a threshold for the influence of HA in brain function [7].
In highlanders living above 4000 m, lower levels of performance in executive functions are observed [8]. Researchers have also measured a reduction in spatial, abstract, and verbal abilities in adolescents living in El Alto, Bolivia (4150 m). HA significantly increases the risk for neurodevelopmental deficits, with larger effects on females [44]. On the contrary, attention deficit hyperactive disorder (ADHD) is less prevalent with increasing altitude, proposing a HA protective effect [45]. Thus, controversy is related to the possible neurocognitive function impairment at HA. However, the corresponding studies conducted so far have the drawback that they do not control biases associated with the socioeconomic level and family, school, and social environments.
In summary, highlanders have adapted to hypoxia through different physiological mechanisms (reviewed in [46]), such as increased lung capacity, increased ventilation, improved oxygen diffusion, increased total vessel density [47], and higher hemoglobin concentration. However, at very HA, impaired cerebral dynamic in blood flow [48], reduced cerebral glucose metabolism [49], reduced cerebral vascular reactivity [50][51], altered gray and white matter [42], and altered neuronal activity are some of the alterations that may affect neurocognitive functioning [52]. Understanding the molecular mechanisms underlying the brain adaptation to HA provides insights into developing interventions to mitigate the adverse effects of HA exposure, such as acute mountain sickness (AMS) and HA cerebral edema (HACE).
2. Hypoxia-Inducible Factors (HIFs) in the Brain Adaptation to HA
At HA, low oxygen supply leads to changes in gene expression mediated by the hypoxia-inducible factors (HIFs) pathway, which play a key role in the cellular response to hypoxia. In 2019, the Nobel Prize in Physiology was awarded to William Kaelin, Peter Ratcliffe, and Gregg Semenza for the discovery of HIFs as master regulators of oxygen homeostasis [53]. HIFs are heterodimers composed of an oxygen-sensitive alpha-subunit (HIF-α) and a constitutive beta-subunit (HIF-1β, also known as ARNT, aryl hydrocarbon receptor nuclear translocator). The two major HIF-α isoforms are HIF-1α and HIF-2α. Although the kinetics of stabilization and transactivation of both isoforms are similar [54], recent genome-wide studies have identified that the EPAS1 gene that encodes for HIF-2α is the major isoform involved in HA adaptation [55]. EPAS1 distribution is tissue- and cell-specific, mainly expressed in organs such as the kidney [56][57] and brain [58].
Hypoxic HIF-2α stabilization at HA leads to the formation of the HIF-2 complex that activates the expression of erythropoietin (EPO). EPO is a glycoprotein hormone that, when released from the kidney, enhances the formation of red blood cells (erythropoiesis) in the bone marrow to increase the blood’s oxygen-carrying capacity [59]. In the brain, EPO regulates the neural respiratory zones (central and peripheral), leading to higher HVR, thereby increasing tissue oxygenation [26]. EPO also increases brain angiogenesis (formation of new vessels) [3][60], neurogenesis (formation of new neurons) [3], synaptogenesis (new synapses) [61], and brain oxidative metabolism and cognition [62][63] (Figure 2).
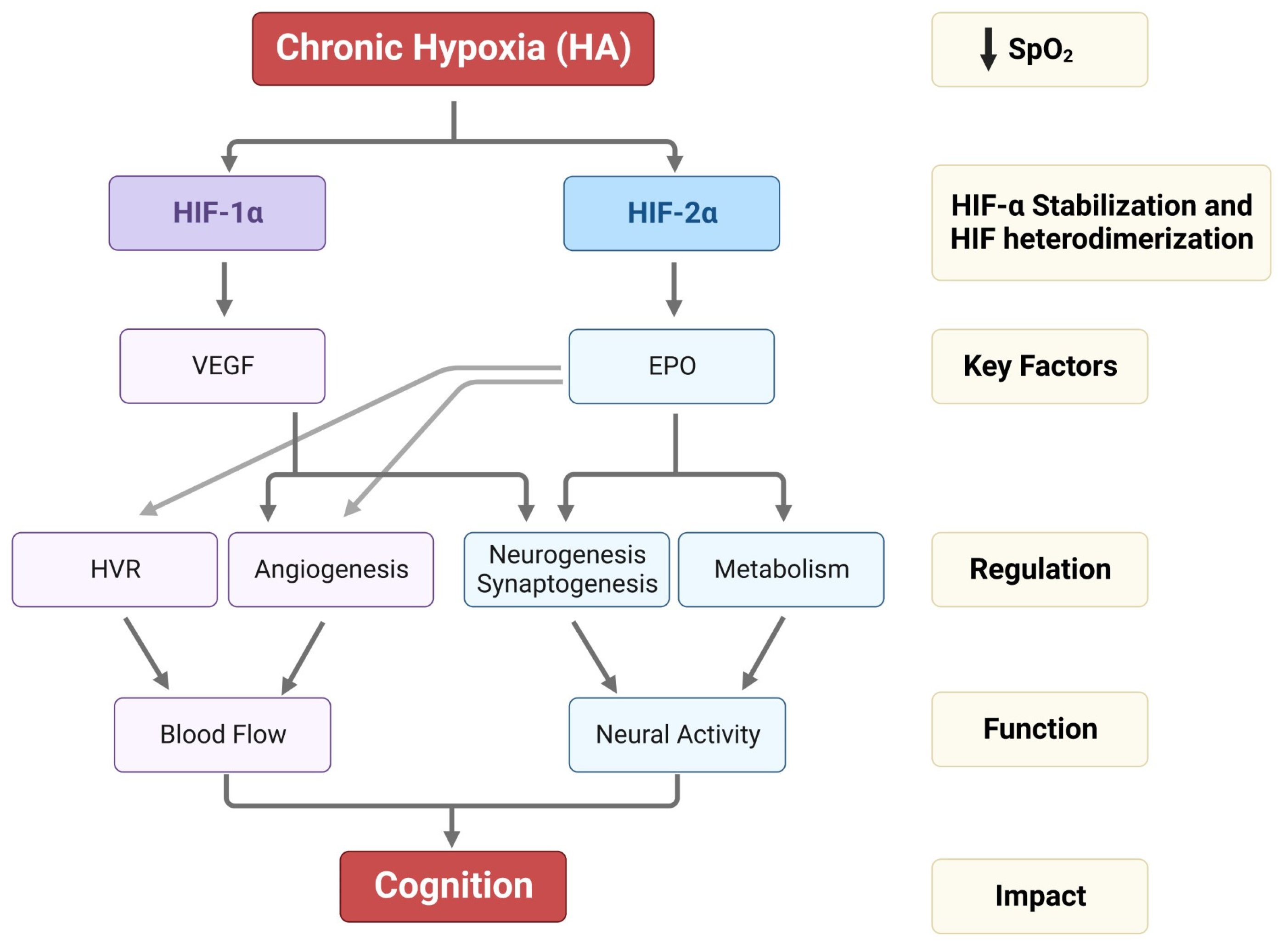
Figure 2. Impact of HA on HIF-1α, HIF-2α stabilization, VEGF and EPO expression, and their regulation in HVR, angiogenesis, neurogenesis, synaptogenesis, and metabolism. The alterations in HVR, angio-neurogenesis, and metabolism influence brain function and cognition.
On the other hand, the HIF-1 complex controls the expression of vascular endothelial growth factor (VEGF), an important signaling protein involved in vasculogenesis, angiogenesis (reviewed in [64]), and neurogenesis [65]. After 11 days of ascent to a HA, researchers observed that inhibition of VEGF signaling in rats interferes with vasculogenesis, angiogenesis, and neurogenesis. In contrast, EPO mainly involves HA-mediated angiogenesis in non-neurogenic brain areas [3] (Figure 2). In Sherpa highlanders, where tolerance to hypoxia is partly attributed to an increase in microcirculatory blood flow and capillary density, the VEGFA plasma levels did not correspond to HA and remained equivalent to the level in non-Sherpa low landers. This finding was speculated to be associated with distinctive genetic variations in the promoter region of VEGFA [66]. Conversely, an increase in VEGFA and EPO in the Andean population is related to chronic mountain sickness (CMS) [67].
HIF-1 also controls the expression of brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-I (IGF-I), both key growth factors that regulate neurogenesis and synaptogenesis from embryonic to adult stages (reviewed in [68][69]). BDNF also interacts with astrocytes and neurons to control respiration [70][71][72]. Animal and human research indicate that physical activity can enhance BDNF blood levels and gene expression [73]. In contrast, long-term exposure to hypoxia lowers BDNF serum levels [74], yet the impact of HA on BDNF expression remains unexplored. No changes in IGF-I expression were observed after acute or chronic exposure to HA [75].
HIF-1 and -2 are crucial for proper brain development and continuously expressed in adult brain neurogenic zones and neural stem/progenitor cells from the embryonic and postnatal mouse brain [76]. HIF-1α deficient mice exhibit hydrocephalus accompanied by reduced neuronal cells and spatial memory impairment [77]. Additionally, HIF-2 has been shown to protect neural progenitor cells and neural differentiation processes by upregulating the survival orthologues Birc5a and Birg5b during embryogenesis [78]. Moreover, recent studies showed that loss of HIF-2α can affect cognitive performance in mice and causes a loss of pyramidal neurons in the retrosplenial cortex, a brain area responsible for spatial navigation [58]. This specific role of HIF-2 in neural development and synaptic plasticity suggests that it is crucial for cognitive function. Thus, targeting HIF-mediated modifications may hold therapeutic potential for HA-related illnesses such as AMS, HACE, and CMS neurologic deficits.
3. Signaling Pathways Interacting with HIFs
Neuronal viability is maintained through a complex interacting network of signaling pathways that can be disturbed in response to many cellular stresses. An imbalance in these signaling pathways after stress or in response to pathology can have drastic consequences for the function or fate of neurons. The mechanisms underlying the HIF-mediated changes in the brain involve a complex interplay between various signaling pathways, including the cyclic adenosine monophosphate (cAMP) pathway, phosphoinositide 3-kinase (PI3K) pathway, and nitric oxide (NO) pathway.
The cAMP pathway is an important second messenger system that plays a critical role in regulating numerous neural processes in the brain, from development, cellular excitability, synaptic plasticity, learning, and memory (reviewed in [79][80][81]). HIF-1 activates the cAMP pathway in response to hypoxia in cancer cells [82][83]. In the brain, cAMP leads to increased expression of the N-methyl-D-aspartate (NMDA) receptor subunit GluN1 in the hippocampus, an area involved in learning and memory [84]. cAMP can also regulate the activity of various downstream effectors involved in synaptic plasticity and memory formation, such as the protein kinase A (PKA) [85] and the cAMP response element-binding protein (CREB) [86]. Moreover, the cAMP signaling pathway can modulate other molecular targets in the brain, such as mitochondrial function and oxidative stress, by activating CREB and the peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) [87]. A longitudinal cohort study of proteomic and clinical biomarkers of AMS symptom phenotypes in 53 individuals after ascent to HA showed that the cAMP pathway interferes with the symptoms of AMS [88]. Therefore, this pathway could be a potential therapeutic strategy for AMS and understanding acclimatization to HA.
The Wnt/β-catenin pathway is directly regulated by HIF and is involved in developing brain structures such as the cerebral cortex and hippocampus (reviewed in [89]). The key mediator of Wnt signaling, the armadillo protein-β-catenin, participates in transcriptional regulation and chromatin interactions and is regulated by hypoxia [89]. The Wnt-mediated activation of HIF-1 promotes the maintenance of embryonic and neural stem cell activity and is significantly decreased in differentiated cells [90]. Furthermore, chronic exposure to hypoxia in vivo induces activation of the Wnt/β-catenin signaling cascade in the hippocampus, suggesting that mild hypoxia may have therapeutic value in neurodegenerative disorders [91].
The PI3K/Akt pathway is another important signaling pathway critical in regulating cell growth and survival and has been widely reported in brain development, aging, neurodegenerative diseases, and psychotic disorders (reviewed in [92]). HIF-1 and PI3K/Akt may interact on functional and regulatory levels. PI3K/Akt is required for heat shock proteins to protect HIF-1α from Von-Hippel-Lindau (pVHL)-independent degradation [93]. However, another earlier study has refuted this dependent interaction [94]. In a rat model of Alzheimer’s disease, it has been shown that gamma-aminobutyric acid (GABA) type B receptor-mediated PI3K/Akt activation alleviates oxidative stress and neuronal cell injury [95]. Moreover, in cerebral ischemic injury, the co-activation of GABAA and GABAB receptors exerted a neuroprotective effect via the PI-3K/Akt pathway [96]. Also, activation of the PI3K/Akt pathway has been shown to have neuroprotective effects and improve cerebral blood flow in animal models of cerebral ischemia [97]. Therefore, targeting this pathway could be a potential therapeutic strategy to treat or prevent HACE. One study demonstrated that treating hypoxic mouse brain microvascular endothelial cells with a PI3K activator, 3-methyladenine, suppressed hypoxia-induced endothelial permeabilization [98] and might effectively tackle HACE. Increased Akt activation (and overexpression of Parkin—a molecule that plays a critical role in ubiquitination) has been shown to reduce hypoxia-induced death of induced pluripotent stem cells-derived neurons from CMS. It is proposed that increased Akt activation protects against hypoxia-induced cell death. Therefore, it is suggested that impaired adaptive mechanisms, including lack of Akt activation and increased Parkin expression, render neurons from chronic mountain sickness subjects more susceptible to hypoxia-induced cell death [99].
The NO signaling pathway plays a significant role in HA-related illnesses, particularly in regulating vascular function and blood flow (reviewed in [100]). Hypoxia-induced HIF activation leads to increased expression of endothelial NO synthase (eNOS) [101]. NO acts as a vasodilator, causing the relaxation of vascular smooth muscle cells, increasing blood flow to oxygen-deprived tissues, and maintaining endothelial homeostasis [102]. NO also plays a pivotal role in regulating neurotransmitter release, such as acetylcholine, catecholamines, and neuroactive amino acids [103], as well as synaptic plasticity in the cerebral cortex [104]. Alterations in NO signaling have been implicated in several HA-related illnesses, including pulmonary edema and HACE [100]. In HA pulmonary edema, impaired NO production and increased pulmonary vasoconstriction can lead to increased fluid accumulation in the lungs. Tibetans have NO levels in the lung, plasma, and red blood cells that are at least double and, in some cases, orders of magnitude greater than in other populations, regardless of altitude [100]. Therapeutic interventions targeting the NO signaling pathway, e.g., administering NO donors or phosphodiesterase inhibitors, have shown promise in preventing and treating HA-related illnesses [105]. HIF-2-regulated EPO induced NO release in endothelial cells [106], cardiomyocytes [107], and lung cancer cells [108]. NO, in turn, co-regulated mitochondria and energy metabolism in combination with Akt. A similar mechanism was witnessed in the murine postnatal hippocampus, corresponding to enhanced cognition in early adulthood [63]. Moreover, NO (and derived S-nitrosothiols and glutathionylation) is required for the physiological response to hypoxia. NO directly interacts with brainstem respiratory centers and modulates the ventilatory response to hypoxia [109]. Furthermore, the current findings suggest that NO’s activity mediates the excitatory and inhibitory components of the hypoxic ventilatory response, and that NO may play a role in modulating the prominent second phase of the biphasic response to hypoxia [110].
Glutathionylation is crucial in redox signaling and cellular adaptation to oxidative stress. It is an important mechanism in regulating the activity of various proteins, including ion transporters and hypoxia-inducible factor 1 (HIF-1) pathway proteins. Sodium/Potassium ATPase (Na+/K+-ATPase) is an essential membrane protein responsible for maintaining the electrochemical gradient across the plasma membrane of cells. Hypoxia induces glutathionylation of specific cysteine residues in the α-subunit of Na+/K+-ATPase. This modification can alter the activity of the pump, affecting ion balance and cellular function [111][112]. Also, glutathionylation of sarcoplasmic/endoplasmic reticulum (ER) calcium ATPase (SERCA) occurs under hypoxia, leading to altered calcium handling and ER stress [113]. Glutathionylation can also affect the DNA-binding activity of HIF-1α. It has been suggested that glutathionylation of specific cysteine residues in HIF-1α may enhance its binding to the hypoxia response elements (HREs) present in the promoter regions of target genes, thereby influencing their transcriptional activation [114]. Glutathionylation of proteins, including ion transporters and HIF-1, is a crucial mechanism in the cellular response to hypoxia. It allows cells to modulate the activity of proteins involved in ion balance, calcium signaling, and gene expression, enabling adaptation and survival under conditions of oxygen limitation. Further research is needed to fully understand the specific cysteine residues targeted for glutathionylation and the functional consequences of this modification on protein activity in the context of hypoxia.
References
- DelMastro, K.; Hellem, T.; Kim, N.; Kondo, D.; Sung, Y.H.; Renshaw, P.F. Incidence of major depressive episode correlates with
- elevation of substate region of residence. J. Affect. Disord. 2011, 129, 376–379. [CrossRef] [PubMed]
- Kious, B.M.; Bakian, A.; Zhao, J.; Mickey, B.; Guille, C.; Renshaw, P.; Sen, S. Altitude and risk of depression and anxiety: Findings
- from the intern health study. Int. Rev. Psychiatry 2019, 31, 637–645. [CrossRef] [PubMed]
- Koester-Hegmann, C.; Bengoetxea, H.; Kosenkov, D.; Thiersch, M.; Haider, T.; Gassmann, M.; Schneider Gasser, E.M. High-
- Altitude Cognitive Impairment Is Prevented by Enriched Environment Including Exercise via VEGF Signaling. Front. Cell.
- Neurosci. 2018, 12, 532. [CrossRef] [PubMed]
- Virues-Ortega, J.; Buela-Casal, G.; Garrido, E.; Alcazar, B. Neuropsychological functioning associated with high-altitude exposure.
- Neuropsychol. Rev. 2004, 14, 197–224. [CrossRef] [PubMed]
- Wang, Y.; Ma, H.; Fu, S.; Guo, S.; Yang, X.; Luo, P.; Han, B. Long-Term Exposure to High Altitude Affects Voluntary Spatial
- Attention at Early and Late Processing Stages. Sci. Rep. 2014, 4, 4443. [CrossRef]
- Bahrke, M.S.; Shukitt-Hale, B. Effects of altitude on mood, behaviour and cognitive functioning. A review. Sports Med. 1993,
- 16, 97–125. [CrossRef]
- Ma, H.; Zhang, D.; Li, X.; Ma, H.; Wang, N.; Wang, Y. Long-term exposure to high altitude attenuates verbal and spatial working
- memory: Evidence from an event-related potential study. Brain Behav. 2019, 9, e01256. [CrossRef]
- Virues-Ortega, J.; Bucks, R.; Kirkham, F.J.; Baldeweg, T.; Baya-Botti, A.; Hogan, A.M.; Bolivian Children Living at Altitude project.
- Changing patterns of neuropsychological functioning in children living at high altitude above and below 4000 m: A report from
- the Bolivian Children Living at Altitude (BoCLA) study. Dev. Sci. 2011, 14, 1185–1193. [CrossRef]
- Shukitt, B.L.; Banderet, L.E. Mood states at 1600 and 4300 meters terrestrial altitude. Aviat. Space Environ. Med. 1988, 59, 530–532.
- Shukitt-Hale, B.; Lieberman, H.R. The Effect of Altitude on Cognitive Performance and Mood States; National Academies Press (US),
- Institute of Medicine (US) Committee on Military Nutrition Research: Washington, DC, USA, 1996; Volume 22.
- Shukitt-Hale, B.; Banderet, L.E.; Lieberman, H.R. Relationships between symptoms, moods, performance, and acute mountain
- sickness at 4700 meters. Aviat. Space Environ. Med. 1991, 62, 865–869.
- Hernandez-Vasquez, A.; Vargas-Fernandez, R.; Rojas-Roque, C.; Gamboa-Unsihuay, J.E. Association between altitude and
- depression in Peru: An 8-year pooled analysis of population-based surveys. J. Affect. Disord. 2022, 299, 536–544. [CrossRef]
- [PubMed]
- Pun, M.; Guadagni, V.; Bettauer, K.M.; Drogos, L.L.; Aitken, J.; Hartmann, S.E.; Furian, M.; Muralt, L.; Lichtblau, M.; Bader, P.R.;
- et al. Effects on Cognitive Functioning of Acute, Subacute and Repeated Exposures to High Altitude. Front. Physiol. 2018, 9, 1131.
- [CrossRef] [PubMed]
- Shukitt-Hale, B.; Stillman, M.J.; Welch, D.I.; Levy, A.; Devine, J.A.; Lieberman, H.R. Hypobaric hypoxia impairs spatial memory
- in an elevation-dependent fashion. Behav. Neural Biol. 1994, 62, 244–252. [CrossRef] [PubMed]
- Drevets, W.C.; Gautier, C.; Price, J.C.; Kupfer, D.J.; Kinahan, P.E.; Grace, A.A.; Price, J.L.; Mathis, C.A. Amphetamine-induced
- dopamine release in human ventral striatum correlates with euphoria. Biol. Psychiatry 2001, 49, 81–96. [CrossRef]
- Young, S.N. Elevated incidence of suicide in people living at altitude, smokers and patients with chronic obstructive pulmonary
- disease and asthma: Possible role of hypoxia causing decreased serotonin synthesis. J. Psychiatry Neurosci. 2013, 38, 423–426.
- [CrossRef]
- Barath, A.S.; Rusheen, A.E.; Rojas Cabrera, J.M.; Price, J.B.; Owen, R.L.; Shin, H.; Jang, D.P.; Blaha, C.D.; Lee, K.H.; Oh, Y.
- Hypoxia-Associated Changes in Striatal Tonic Dopamine Release: Real-Time in vivo Measurements with a Novel Voltammetry
- Technique. Front. Neurosci. 2020, 14, 869. [CrossRef]
- Huang, C.C.; Lajevardi, N.S.; Tammela, O.; Pastuszko, A.; Delivoria-Papadopoulos, M.; Wilson, D.F. Relationship of extracellular
- dopamine in striatum of newborn piglets to cortical oxygen pressure. Neurochem. Res. 1994, 19, 649–655. [CrossRef]
- Pitychoutis, P.M.; Dalla, C.; Sideris, A.C.; Tsonis, P.A.; Papadopoulou-Daifoti, Z. 5-HT(1A), 5-HT(2A), and 5-HT(2C) receptor
- mRNA modulation by antidepressant treatment in the chronic mild stress model of depression: Sex differences exposed.
- Neuroscience 2012, 210, 152–167. [CrossRef]
- Ma, S.; Mifflin, S.W.; Cunningham, J.T.; Morilak, D.A. Chronic intermittent hypoxia sensitizes acute hypothalamic-pituitary-
- adrenal stress reactivity and Fos induction in the rat locus coeruleus in response to subsequent immobilization stress. Neuroscience
- 2008, 154, 1639–1647. [CrossRef]
- Kondo, D.G.; Forrest, L.N.; Shi, X.; Sung, Y.H.; Hellem, T.L.; Huber, R.S.; Renshaw, P.F. Creatine target engagement with brain
- bioenergetics: A dose-ranging phosphorus-31 magnetic resonance spectroscopy study of adolescent females with SSRI-resistant
- depression. Amino Acids 2016, 48, 1941–1954. [CrossRef]
- Jain, K.; Prasad, D.; Singh, S.B.; Kohli, E. Hypobaric Hypoxia Imbalances Mitochondrial Dynamics in Rat Brain Hippocampus.
- Neurol. Res. Int. 2015, 2015, 742059. [CrossRef]
- Shi, X.F.; Carlson, P.J.; Kim, T.S.; Sung, Y.H.; Hellem, T.L.; Fiedler, K.K.; Kim, S.E.; Glaeser, B.; Wang, K.; Zuo, C.S.; et al. Effect of
- altitude on brain intracellular pH and inorganic phosphate levels. Psychiatry Res. 2014, 222, 149–156. [CrossRef] [PubMed]
- Penaloza, D.; Arias-Stella, J. The heart and pulmonary circulation at high altitudes: Healthy highlanders and chronic mountain
- sickness. Circulation 2007, 115, 1132–1146. [CrossRef] [PubMed]
- Rojas-Camayo, J.; Mejia, C.R.; Callacondo, D.; Dawson, J.A.; Posso, M.; Galvan, C.A.; Davila-Arango, N.; Bravo, E.A.; Loescher,
- V.Y.; Padilla-Deza, M.M.; et al. Reference values for oxygen saturation from sea level to the highest human habitation in the
- Andes in acclimatised persons. Thorax 2018, 73, 776–778. [CrossRef]
- Soliz, J.; Joseph, V.; Soulage, C.; Becskei, C.; Vogel, J.; Pequignot, J.M.; Ogunshola, O.; Gassmann, M. Erythropoietin regulates
- hypoxic ventilation in mice by interacting with brainstem and carotid bodies. J. Physiol. 2005, 568, 559–571. [CrossRef]
- Lombardi, C.; Meriggi, P.; Agostoni, P.; Faini, A.; Bilo, G.; Revera, M.; Caldara, G.; Di Rienzo, M.; Castiglioni, P.; Maurizio, B.; et al.
- High-altitude hypoxia and periodic breathing during sleep: Gender-related differences. J. Sleep. Res. 2013, 22, 322–330. [CrossRef]
- Julian, C.G.; Vargas, E.; Gonzales, M.; Davila, R.D.; Ladenburger, A.; Reardon, L.; Schoo, C.; Powers, R.W.; Lee-Chiong, T.;
- Moore, L.G. Sleep-disordered breathing and oxidative stress in preclinical chronic mountain sickness (excessive erythrocytosis).
- Respir. Physiol. Neurobiol. 2013, 186, 188–196. [CrossRef]
- Beall, C.M. Oxygen saturation increases during childhood and decreases during adulthood among high altitude native Tibetians
- residing at 3800–4200 m. High. Alt. Med. Biol. 2000, 1, 25–32. [CrossRef]
- Hill, C.M.; Baya, A.; Gavlak, J.; Carroll, A.; Heathcote, K.; Dimitriou, D.; L’Esperance, V.; Webster, R.; Holloway, J.;
- Virues-Ortega, J.; et al. Adaptation to Life in the High Andes: Nocturnal Oxyhemoglobin Saturation in Early Development. Sleep
- 2016, 39, 1001–1008. [CrossRef]
- Ucros, S.; Granados, C.M.; Castro-Rodriguez, J.A.; Hill, C.M. Oxygen Saturation in Childhood at High Altitude: A Systematic
- Review. High. Alt. Med. Biol. 2020, 21, 114–125. [CrossRef]
- Ucros, S.; Castro-Guevara, J.A.; Hill, C.M.; Castro-Rodriguez, J.A. Breathing Patterns and Oxygenation Saturation during Sleep
- in Children Habitually Living at High Altitude in the Andes: A Systematic Review. Front. Pediatr. 2021, 9, 798310. [CrossRef]
- Ucros, S.; Granados, C.; Hill, C.; Castro-Rodriguez, J.A.; Ospina, J.C. Normal values for respiratory sleep polygraphy in children
- aged 4 to 9 years at 2560 m above sea level. J. Sleep. Res. 2021, 30, e13341. [CrossRef] [PubMed]
- de Aquino Lemos, V.; Antunes, H.K.; dos Santos, R.V.; Lira, F.S.; Tufik, S.; de Mello, M.T. High altitude exposure impairs sleep
- patterns, mood, and cognitive functions. Psychophysiology 2012, 49, 1298–1306. [CrossRef] [PubMed]
- Nation, D.A.; Bondi, M.W.; Gayles, E.; Delis, D.C. Mechanisms of Memory Dysfunction during High Altitude Hypoxia Training
- in Military Aircrew. J. Int. Neuropsychol. Soc. 2017, 23, 1–10. [CrossRef] [PubMed]
- Pavlicek, V.; Schirlo, C.; Nebel, A.; Regard, M.; Koller, E.A.; Brugger, P. Cognitive and emotional processing at high altitude. Aviat.
- Space Environ. Med. 2005, 76, 28–33.
- Zhang, D.; Ma, H.; Huang, J.; Zhang, X.; Ma, H.; Liu, M. Exploring the impact of chronic high-altitude exposure on visual spatial
- attention using the ERP approach. Brain Behav. 2018, 8, e00944. [CrossRef]
- Fayed, N.; Modrego, P.J.; Morales, H. Evidence of brain damage after high-altitude climbing by means of magnetic resonance
- imaging. Am. J. Med. 2006, 119, 168.e1–168.e6. [CrossRef]
- Fan, C.; Zhao, Y.; Yu, Q.; Yin, W.; Liu, H.; Lin, J.; Yang, T.; Fan, M.; Gesang, L.; Zhang, J. Reversible Brain Abnormalities in People
- without Signs of Mountain Sickness during High-Altitude Exposure. Sci. Rep. 2016, 6, 33596. [CrossRef]
- Zhang, H.; Lin, J.; Sun, Y.; Huang, Y.; Ye, H.; Wang, X.; Yang, T.; Jiang, X.; Zhang, J. Compromised white matter microstructural
- integrity after mountain climbing: Evidence from diffusion tensor imaging. High. Alt. Med. Biol. 2012, 13, 118–125. [CrossRef]
- Chen, J.; Li, J.; Han, Q.; Lin, J.; Yang, T.; Chen, Z.; Zhang, J. Long-term acclimatization to high-altitude hypoxia modifies
- interhemispheric functional and structural connectivity in the adult brain. Brain Behav. 2016, 6, e00512. [CrossRef]
- Zhang, X.; Zhang, J. The human brain in a high altitude natural environment: A review. Front. Hum. Neurosci. 2022, 16, 915995.
- Richardson, C.; Hogan, A.M.; Bucks, R.S.; Baya, A.; Virues-Ortega, J.; Holloway, J.W.; Rose-Zerilli, M.; Palmer, L.J.; Webster, R.J.;
- Kirkham, F.J.; et al. Neurophysiological evidence for cognitive and brain functional adaptation in adolescents living at high
- altitude. Clin. Neurophysiol. 2011, 122, 1726–1734. [CrossRef] [PubMed]
- Wehby, G.L. Living on higher ground reduces child neurodevelopment-evidence from South America. J. Pediatr. 2013,
- 162, 606–611.e1. [CrossRef] [PubMed]
- Huber, R.S.; Kim, T.S.; Kim, N.; Kuykendall, M.D.; Sherwood, S.N.; Renshaw, P.F.; Kondo, D.G. Association between Altitude and
- Regional Variation of ADHD in Youth. J. Atten. Disord. 2018, 22, 1299–1306. [CrossRef]
- Mallet, R.T.; Burtscher, J.; Pialoux, V.; Pasha, Q.; Ahmad, Y.; Millet, G.P.; Burtscher, M. Molecular Mechanisms of High-Altitude
- Acclimatization. Int. J. Mol. Sci. 2023, 24, 1968. [CrossRef]
- Gassmann, N.N.; van Elteren, H.A.; Goos, T.G.; Morales, C.R.; Rivera-Ch, M.; Martin, D.S.; Peralta, P.C.; Passano Del Carpio, A.;
- Aranibar Machaca, S.; Huicho, L.; et al. Pregnancy at high altitude in the Andes leads to increased total vessel density in healthy
- newborns. J. Appl. Physiol. 2016, 121, 709–715. [CrossRef]
- Iwasaki, K.; Zhang, R.; Zuckerman, J.H.; Ogawa, Y.; Hansen, L.H.; Levine, B.D. Impaired dynamic cerebral autoregulation at
- extreme high altitude even after acclimatization. J. Cereb. Blood Flow. Metab. 2011, 31, 283–292. [CrossRef]
- Merz, T.M.; Treyer, V.; Hefti, U.; Spengler, C.M.; Schwarz, U.; Buck, A.; Maggiorini, M. Changes in cerebral glucose metabolism
- after an expedition to high altitudes. High. Alt. Med. Biol. 2006, 7, 28–38. [CrossRef]
- Hogan, A.M.; Virues-Ortega, J.; Botti, A.B.; Bucks, R.; Holloway, J.W.; Rose-Zerilli, M.J.; Palmer, L.J.; Webster, R.J.; Baldeweg, T.;
- Kirkham, F.J. Development of aptitude at altitude. Dev. Sci. 2010, 13, 533–544. [CrossRef]
- Yan, X. Cognitive impairments at high altitudes and adaptation. High Alt. Med. Biol. 2014, 15, 141–145. [CrossRef]
- Chen, X.; Liu, J.; Wang, J.; Xin, Z.; Zhang, Q.; Zhang, W.; Xi, Y.; Zhu, Y.; Li, C.; Li, J.; et al. Altered resting-state networks may
- explain the executive impairment in young health immigrants into high-altitude area. Brain Imaging Behav. 2021, 15, 147–156.
- Ledford, H.; Callaway, E. Biologists who decoded how cells sense oxygen win medicine Nobel. Nature 2019, 574, 161–162.
- Bracken, C.P.; Fedele, A.O.; Linke, S.; Balrak, W.; Lisy, K.; Whitelaw, M.L.; Peet, D.J. Cell-specific regulation of hypoxia-inducible
- factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J. Biol. Chem. 2006,
- 281, 22575–22585. [CrossRef]
- Li, C.; Li, X.; Xiao, J.; Liu, J.; Fan, X.; Fan, F.; Lei, H. Genetic changes in the EPAS1 gene between Tibetan and Han ethnic groups
- and adaptation to the plateau hypoxic environment. PeerJ 2019, 7, e7943. [CrossRef] [PubMed]
- Paliege, A.; Rosenberger, C.; Bondke, A.; Sciesielski, L.; Shina, A.; Heyman, S.N.; Flippin, L.A.; Arend, M.; Klaus, S.J.; Bachmann,
- S. Hypoxia-inducible factor-2alpha-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under
- hypoxia-inducible factor stabilization. Kidney Int. 2010, 77, 312–318. [CrossRef] [PubMed]
- Lee, F.S.; Percy, M.J. The HIF pathway and erythrocytosis. Annu. Rev. Pathol. 2011, 6, 165–192. [CrossRef] [PubMed]
- Kleszka, K.; Leu, T.; Quinting, T.; Jastrow, H.; Pechlivanis, S.; Fandrey, J.; Schreiber, T. Hypoxia-inducible factor-2alpha is crucial
- for proper brain development. Sci. Rep. 2020, 10, 19146. [CrossRef]
- Jacobson, L.O.; Goldwasser, E.; Fried, W.; Plzak, L.F. Studies on erythropoiesis. VII. The role of the kidney in the production of
- erythropoietin. Trans. Assoc. Am. Physicians 1957, 70, 305–317.
- Kimakova, P.; Solar, P.; Solarova, Z.; Komel, R.; Debeljak, N. Erythropoietin and Its Angiogenic Activity. Int. J. Mol. Sci. 2017, 18, 1519.
- Khalid, K.; Frei, J.; Aboouf, M.A.; Koester-Hegmann, C.; Gassmann, M.; Fritschy, J.M.; Schneider Gasser, E.M. Erythropoietin
- Stimulates GABAergic Maturation in the Mouse Hippocampus. eNeuro 2021, 8, 0006–0021. [CrossRef]
- Ehrenreich, H.; Garcia-Agudo, L.F.; Steixner-Kumar, A.A.; Wilke, J.B.H.; Butt, U.J. Introducing the brain erythropoietin circle to
- explain adaptive brain hardware upgrade and improved performance. Mol. Psychiatry 2022, 27, 2372–2379. [CrossRef] [PubMed]
- Jacobs, R.A.; Aboouf, M.A.; Koester-Hegmann, C.; Muttathukunnel, P.; Laouafa, S.; Arias-Reyes, C.; Thiersch, M.; Soliz, J.;
- Gassmann, M.; Schneider Gasser, E.M. Erythropoietin promotes hippocampal mitochondrial function and enhances cognition in
- mice. Commun. Biol. 2021, 4, 938. [CrossRef] [PubMed]
- Fong, G.H. Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis 2008, 11, 121–140. [CrossRef] [PubMed]
- Jin, K.; Zhu, Y.; Sun, Y.; Mao, X.O.; Xie, L.; Greenberg, D.A. Vascular endothelial growth factor (VEGF) stimulates neurogenesis
- in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11946–11950. [CrossRef]
- Droma, Y.; Hanaoka, M.; Kinjo, T.; Kobayashi, N.; Yasuo, M.; Kitaguchi, Y.; Ota, M. The blunted vascular endothelial growth
- factor-A (VEGF-A) response to high-altitude hypoxia and genetic variants in the promoter region of the VEGFA gene in Sherpa
- highlanders. PeerJ 2022, 10, e13893. [CrossRef]
- Espinoza, J.R.; Alvarez, G.; Leon-Velarde, F.; Preciado, H.F.; Macarlupu, J.L.; Rivera-Ch, M.; Rodriguez, J.; Favier, J.; Gimenez-
- Roqueplo, A.P.; Richalet, J.P. Vascular endothelial growth factor-A is associated with chronic mountain sickness in the Andean
- population. High Alt. Med. Biol. 2014, 15, 146–154. [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the
- Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [CrossRef]
- Nieto-Estevez, V.; Defterali, C.; Vicario-Abejon, C. IGF-I: A Key Growth Factor that Regulates Neurogenesis and Synaptogenesis
- from Embryonic to Adult Stages of the Brain. Front. Neurosci. 2016, 10, 52. [CrossRef]
- Balkowiec, A.; Katz, D.M. Brain-derived neurotrophic factor is required for normal development of the central respiratory rhythm
- in mice. J. Physiol. 1998, 510, 527–533. [CrossRef]
- Erickson, J.T.; Conover, J.C.; Borday, V.; Champagnat, J.; Barbacid, M.; Yancopoulos, G.; Katz, D.M. Mice lacking brain-derived
- neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental
- deficit in control of breathing. J. Neurosci. 1996, 16, 5361–5371. [CrossRef]
- Caravagna, C.; Soliz, J.; Seaborn, T. Brain-derived neurotrophic factor interacts with astrocytes and neurons to control respiration.
- Eur. J. Neurosci. 2013, 38, 3261–3269. [CrossRef] [PubMed]
- Enette, L.; Vogel, T.; Fanon, J.L.; Lang, P.O. Effect of Interval and Continuous Aerobic Training on Basal Serum and Plasma Brain-
- Derived Neurotrophic Factor Values in Seniors: A Systematic Review of Intervention Studies. Rejuvenation Res. 2017, 20, 473–483.
- Becke, A.; Muller, P.; Dordevic, M.; Lessmann, V.; Brigadski, T.; Muller, N.G. Daily Intermittent Normobaric Hypoxia Over 2
- Weeks Reduces BDNF Plasma Levels in Young Adults—A Randomized Controlled Feasibility Study. Front. Physiol. 2018, 9, 1337.
- Richalet, J.P.; Letournel, M.; Souberbielle, J.C. Effects of high-altitude hypoxia on the hormonal response to hypothalamic factors.
- Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1685–R1692. [CrossRef]
- Roitbak, T.; Surviladze, Z.; Cunningham, L.A. Continuous expression of HIF-1alpha in neural stem/progenitor cells. Cell. Mol.
- Neurobiol. 2011, 31, 119–133. [CrossRef]
- Tomita, S.; Ueno, M.; Sakamoto, M.; Kitahama, Y.; Ueki, M.; Maekawa, N.; Sakamoto, H.; Gassmann, M.; Kageyama, R.; Ueda,
- N.; et al. Defective brain development in mice lacking the Hif-1alpha gene in neural cells. Mol. Cell. Biol. 2003, 23, 6739–6749.
- Ko, C.Y.; Tsai, M.Y.; Tseng, W.F.; Cheng, C.H.; Huang, C.R.; Wu, J.S.; Chung, H.Y.; Hsieh, C.S.; Sun, C.K.; Hwang, S.P.; et al.
- Integration of CNS survival and differentiation by HIF2alpha. Cell. Death Differ. 2011, 18, 1757–1770. [CrossRef] [PubMed]
- Pierre, S.; Eschenhagen, T.; Geisslinger, G.; Scholich, K. Capturing adenylyl cyclases as potential drug targets. Nat. Rev. Drug.
- Discov. 2009, 8, 321–335. [CrossRef]
- Kandel, E.R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 2012, 5, 14. [CrossRef]
- Bollen, E.; Prickaerts, J. Phosphodiesterases in neurodegenerative disorders. IUBMB Life 2012, 64, 965–970. [CrossRef]
- Seaayfan, E.; Nasrah, S.; Quell, L.; Radi, A.; Kleim, M.; Schermuly, R.T.; Weber, S.; Laghmani, K.; Komhoff, M. Reciprocal
- Regulation of MAGED2 and HIF-1alpha Augments Their Expression under Hypoxia: Role of cAMP and PKA Type II. Cells 2022,
- 11, 3424. [CrossRef] [PubMed]
- Simko, V.; Iuliano, F.; Sevcikova, A.; Labudova, M.; Barathova, M.; Radvak, P.; Pastorekova, S.; Pastorek, J.; Csaderova, L. Hypoxia
- induces cancer-associated cAMP/PKA signalling through HIF-mediated transcriptional control of adenylyl cyclases VI and VII.
- Sci. Rep. 2017, 7, 10121. [CrossRef] [PubMed]
- Haddad, J.J. The bioanalytical molecular pharmacology of the N-methyl-D-aspartate (NMDA) receptor nexus and the oxygen-
- responsive transcription factor HIF-1alpha: Putative mechanisms and regulatory pathways unravel the intimate hypoxia
- connection. Curr. Mol. Pharmacol. 2013, 6, 104–135. [CrossRef] [PubMed]
- Shahoha, M.; Cohen, R.; Ben-Simon, Y.; Ashery, U. cAMP-Dependent Synaptic Plasticity at the Hippocampal Mossy Fiber
- Terminal. Front. Synaptic Neurosci. 2022, 14, 861215. [CrossRef]
- Benito, E.; Barco, A. CREB’s control of intrinsic and synaptic plasticity: Implications for CREB-dependent memory models. Trends
- Neurosci. 2010, 33, 230–240. [CrossRef]
- Signorile, A.; De Rasmo, D. Mitochondrial Complex I, a Possible Sensible Site of cAMP Pathway in Aging. Antioxidants 2023, 12, 221.
- Yang, J.; Jia, Z.; Song, X.; Shi, J.; Wang, X.; Zhao, X.; He, K. Proteomic and clinical biomarkers for acute mountain sickness in a
- longitudinal cohort. Commun. Biol. 2022, 5, 548. [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell. Dev. Biol. 2004, 20, 781–810.
- Pouyssegur, J.; Lenormand, P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Eur. J. Biochem. 2003,
- 270, 3291–3299. [CrossRef]
- Varela-Nallar, L.; Rojas-Abalos, M.; Abbott, A.C.; Moya, E.A.; Iturriaga, R.; Inestrosa, N.C. Chronic hypoxia induces the activation
- of the Wnt/beta-catenin signaling pathway and stimulates hippocampal neurogenesis in wild-type and APPswe-PS1DeltaE9
- transgenic mice in vivo. Front. Cell. Neurosci. 2014, 8, 17. [CrossRef]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med.
- Rep. 2018, 18, 3547–3554. [CrossRef] [PubMed]
- Zhou, J.; Schmid, T.; Frank, R.; Brune, B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1alpha
- from pVHL-independent degradation. J. Biol. Chem. 2004, 279, 13506–13513. [CrossRef] [PubMed]
- Arsham, A.M.; Plas, D.R.; Thompson, C.B.; Simon, M.C. Phosphatidylinositol 3-kinase/Akt signaling is neither required
- for hypoxic stabilization of HIF-1 alpha nor sufficient for HIF-1-dependent target gene transcription. J. Biol. Chem. 2002,
- 277, 15162–15170. [CrossRef] [PubMed]
- Sun, Z.; Sun, L.; Tu, L. GABAB Receptor-Mediated PI3K/Akt Signaling Pathway Alleviates Oxidative Stress and Neuronal Cell
- Injury in a Rat Model of Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1513–1526. [CrossRef] [PubMed]
- Xu, J.; Li, C.; Yin, X.H.; Zhang, G.Y. Additive neuroprotection of GABA A and GABA B receptor agonists in cerebral ischemic
- injury via PI-3K/Akt pathway inhibiting the ASK1-JNK cascade. Neuropharmacology 2008, 54, 1029–1040. [CrossRef]
- Lv, M.R.; Li, B.; Wang, M.G.; Meng, F.G.; Yu, J.J.; Guo, F.; Li, Y. Activation of the PI3K-Akt pathway promotes neuroprotection of
- the delta-opioid receptor agonist against cerebral ischemia-reperfusion injury in rat models. Biomed. Pharmacother. 2017, 93, 230–237.
- Xue, Y.; Wang, X.; Wan, B.; Wang, D.; Li, M.; Cheng, K.; Luo, Q.; Wang, D.; Lu, Y.; Zhu, L. Caveolin-1 accelerates hypoxia-induced
- endothelial dysfunction in high-altitude cerebral edema. Cell. Commun. Signal. 2022, 20, 160. [CrossRef] [PubMed]
- Zhao, H.; Lin, J.; Sieck, G.; Haddad, G.G. Neuroprotective Role of Akt in Hypoxia Adaptation in Andeans. Front. Neurosci. 2020,
- 14, 607711. [CrossRef] [PubMed]
- Beall, C.M.; Laskowski, D.; Erzurum, S.C. Nitric oxide in adaptation to altitude. Free. Radic. Biol. Med. 2012, 52, 1123–1134.
- Rodriguez-Miguelez, P.; Lima-Cabello, E.; Martinez-Florez, S.; Almar, M.; Cuevas, M.J.; Gonzalez-Gallego, J. Hypoxia-inducible
- factor-1 modulates the expression of vascular endothelial growth factor and endothelial nitric oxide synthase induced by eccentric
- exercise. J. Appl. Physiol. 2015, 118, 1075–1083. [CrossRef]
- Heiss, C.; Rodriguez-Mateos, A.; Kelm, M. Central role of eNOS in the maintenance of endothelial homeostasis. Antioxid. Redox
- Signal. 2015, 22, 1230–1242. [CrossRef]
- Kuriyama, K.; Ohkuma, S. Role of nitric oxide in central synaptic transmission: Effects on neurotransmitter release. Jpn. J.
- Pharmacol. 1995, 69, 1–8. [CrossRef] [PubMed]
- Hardingham, N.; Dachtler, J.; Fox, K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front. Cell. Neurosci. 2013,
- 7, 190. [CrossRef] [PubMed]
- Scherrer, U.; Vollenweider, L.; Delabays, A.; Savcic, M.; Eichenberger, U.; Kleger, G.R.; Fikrle, A.; Ballmer, P.E.; Nicod, P.; Bartsch,
- P. Inhaled nitric oxide for high-altitude pulmonary edema. N. Engl. J. Med. 1996, 334, 624–629. [CrossRef] [PubMed]
- Beleslin-Cokic, B.B.; Cokic, V.P.; Yu, X.; Weksler, B.B.; Schechter, A.N.; Noguchi, C.T. Erythropoietin and hypoxia stimulate
- erythropoietin receptor and nitric oxide production by endothelial cells. Blood 2004, 104, 2073–2080. [CrossRef] [PubMed]
- Carraway, M.S.; Suliman, H.B.; Jones, W.S.; Chen, C.W.; Babiker, A.; Piantadosi, C.A. Erythropoietin activates mitochondrial
- biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ. Res. 2010, 106, 1722–1730. [CrossRef]
- Aboouf, M.A.; Guscetti, F.; von Buren, N.; Armbruster, J.; Ademi, H.; Ruetten, M.; Melendez-Rodriguez, F.; Rulicke, T.; Seymer,
- A.; Jacobs, R.A.; et al. Erythropoietin receptor regulates tumor mitochondrial biogenesis through iNOS and pAKT. Front. Oncol.
- 2022, 12, 976961. [CrossRef]
- Lipton, A.J.; Johnson, M.A.; Macdonald, T.; Lieberman, M.W.; Gozal, D.; Gaston, B. S-nitrosothiols signal the ventilatory response
- to hypoxia. Nature 2001, 413, 171–174. [CrossRef]
- Gozal, D.; Gozal, E.; Torres, J.E.; Gozal, Y.M.; Nuckton, T.J.; Hornby, P.J. Nitric oxide modulates ventilatory responses to hypoxia
- in the developing rat. Am. J. Respir. Crit. Care Med. 1997, 155, 1755–1762. [CrossRef]
- Comellas, A.P.; Dada, L.A.; Lecuona, E.; Pesce, L.M.; Chandel, N.S.; Quesada, N.; Budinger, G.R.; Strous, G.J.; Ciechanover,
- A.; Sznajder, J.I. Hypoxia-mediated degradation of Na,K-ATPase via mitochondrial reactive oxygen species and the ubiquitin-
- conjugating system. Circ. Res. 2006, 98, 1314–1322. [CrossRef]
- Mitkevich, V.A.; Petrushanko, I.Y.; Poluektov, Y.M.; Burnysheva, K.M.; Lakunina, V.A.; Anashkina, A.A.; Makarov, A.A. Basal
- Glutathionylation of Na,K-ATPase alpha-Subunit Depends on Redox Status of Cells during the Enzyme Biosynthesis. Oxid. Med.
- Cell. Longev. 2016, 2016, 9092328. [CrossRef] [PubMed]
- Thompson, M.D.; Mei, Y.; Weisbrod, R.M.; Silver, M.; Shukla, P.C.; Bolotina, V.M.; Cohen, R.A.; Tong, X. Glutathione adducts on
- sarcoplasmic/endoplasmic reticulum Ca2+ ATPase Cys-674 regulate endothelial cell calcium stores and angiogenic function as
- well as promote ischemic blood flow recovery. J. Biol. Chem. 2014, 289, 19907–19916. [CrossRef]
- Jiang, B.H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA binding, and transactivation properties of hypoxia-
- inducible factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [CrossRef] [PubMed]
More
Information
Subjects:
Physiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Revisions:
3 times
(View History)
Update Date:
29 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No