Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Edith Marianne Schneider Gasser and Version 3 by Markus Thiersch.
The brain requires over one-fifth of the total body oxygen demand for normal functioning. At high altitude (HA), the lower atmospheric oxygen pressure inevitably challenges the brain, affecting voluntary spatial attention, cognitive processing, and attention speed after short-term, long-term, or lifespan exposure. Molecular responses to HA are controlled mainly by hypoxia-inducible factors.
- hypoxia
- HIF
- EPO
- high altitude
1. High Altitude and Cognition
High altitude (HA) (defined as an altitude above 2500 m above sea level) is characterized by multiple harsh environmental conditions. Most physiological adaptations occur in response to the reduced atmospheric pressure, resulting in reduced oxygen partial pressure and causing reduced blood oxygenation saturation (SpO2) hypoxemia. The brain is susceptible to alterations in oxygen supply. Thus, HA exposure causes adverse changes in mood states, such as depression [1] and anxiety [2], and neurocognitive alterations, such as memory impairment [3] and attention disorders after both short- and long-term HA exposure [4][5]. Although numerous reports concern the physiological and neurological alteration that occurs after ascent to HA, less has been investigated on the cognitive and brain alterations in long-term and permanent inhabitants of HA.
Brain functions are affected by hypoxia not only after ascent to HA [6] but also after long-term exposure at a HA [7] and in native highlanders [8]. In unacclimatized individuals exposed to HA, sleep patterns can already be affected at elevations above 1600 m, changes in mood states like euphoria or depression are observed in some individuals from 2500 m on, and above 3000 m subjects can experience headache, dizziness, and confusion. Mood state alterations, including euphoria, quarrelsome, irritability, and apathy, occur temporarily after fast acute exposure to HA and return to baseline states after 48 to 52 h [9][10][11]. In contrast, short- and long-term exposure to HA causes biological, inflammatory, and structural brain changes that increase the risk of experiencing anxiety and depression symptoms [12] and neurocognitive dysfunctions such as slower reaction times, reduced attention (>3500 m), impaired learning, spatial and working memory (>4000 m), and impaired retrieval (>5500 m) (Figure 1) [7][8][13][14].
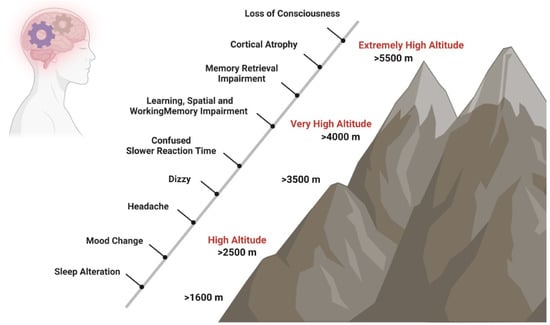
Figure 1. Impact of HA on cognition. Short- and long-term exposure to HA has effects on neurocognitive functions. Sleep patterns can be affected at elevations above 1600 m, changes in mood states like euphoria or depression are observed in some individuals from 2500 m on, and above 3000 m, subjects can experience headache, dizziness, and confusion. At very HA, slower reaction times, such as reduced attention (>3500 m), impaired learning, spatial and working memory (>4000 m), and impaired retrieval (>5500 m) may occur.
Mood alterations after HA exposure are attributed to changes in brain levels of dopamine and serotonin. Euphoria is attributed to increased brain dopamine levels [15], while lower serotonin levels are attributed to sadness, grief, worry, anxiety, and depression [16]. Studies in rodents and piglets have shown increased extracellular dopamine levels in the striatum when the oxygen pressure in the cerebral cortex decreases [17][18]. Moreover, reduced serotonin (5-hydroxytryptamine, 5-HT) availability is observed at HA in people with anxiety and depression, particularly in women [19]. Long-term exposure to HA induces a hypoxic stress response in which the noradrenergic brain regions, particularly the locus coeruleus and hypothalamic-pituitary-adrenal (HPA) axis, are involved [20]. Similarly, HA leads to alterations in brain bioenergetics, which could also contribute to depressive symptoms. Indeed, multiple magnetic resonance spectroscopy studies suggest that persons at HA with depression exhibit alteration in adenosine triphosphate (ATP) expression [21]. Similarly, an imbalance in mitochondrial dynamics has been demonstrated in the rat brain hippocampus after hypobaric hypoxia exposure [22]. Moreover, HA residents (1400 m, Salt Lake City in Utah, USA) exhibit significant differences in brain pH and inorganic phosphate levels compared with sea-level residents (13 m, Belmont City in Massachusetts, USA) [23].
Acute, subacute, and repeated HA exposure causes a reduction in neurocognitive processing speed (reaction time) and attention (switching, visual processing). A study in healthy subjects showed that acclimatization improves reaction time and attention due to increased SpO2. Thus, processing speed and attention are associated with SpO2 acclimatization [13].
Despite about 2.2% of the world’s population (more than 140 million people) living permanently at a HA [24], the impact of reduced SpO2 on cognition remains poorly explored. SpO2 values decrease with increasing altitude to a median of 96% (95–97) at 2500 m, 92% (90–93) at 3600 m, 87% (85–89) at 4100, and 81% (78–84) at 5100 m, with increasing variability at higher altitudes [25]. The reduced SpO2 at HAs induces a rapid increase in hypoxic ventilatory response (HVR) [26], but also breathing instability (periods of deep and rapid breathing) and central apnea during sleep [27]. Although the rate of sleep apnea is lower at HA than at sea level [28], the gap in SpO2 between wakefulness and sleep states is greater than at sea level [29][30]. The statistical distribution of SpO2 in children at HA shows that one out of four children saturates significantly less than the others [31]. Moreover, in one out of four children at HA, the oxygen desaturation index during sleep (which reflects SpO2 variability) is lower [32][33]. Changes in sleep patterns can modulate mood and cause deficits in working memory, attention, or executive functions [34]. Yet, whether low SpO2 during sleep correlates with impaired cognition at HA remains unknown.
Acute exposure to HA causes impairment in learning and memory encoding and retention [35]. Those alterations are also observed in lowlanders living at HA [36]. With the development of cognitive neuroscience research techniques such as functional magnetic resonance imaging (fMRI) and event-related potentials (ERP), it is possible now to explore the mechanisms underlying the effects of long-term HA exposure on cognition. After natural selection, highlanders have genetic and physiological adaptations to HA. Thus, brain alterations in HA-exposed lowlanders may not reflect the brain of adapted highlanders. Exposure to HA affects mainly visual spatial attention in lowlanders [37]. After the ascent to very HA, brain edema and cortical atrophy were reported in climbers [38], but the reduction in brain volume was only reported after more than three weeks of very HA exposure (>5500 m) [39]. For HA immigrants, an increase in gray matter is observed, probably associated with increased neurogenesis and vasculature. Alterations in white matter volumes after acute HA exposure were rarely observed, but diffuse imaging suggested injury [40]. Highlander descendants of Han immigrants showed increased interhemispheric fibers [41]. The changes in cerebrospinal fluid were very variable across studies [42].
fMRI is used to measure changes in blood dynamics caused by neuronal activity. Blood oxygenation level-dependent (BOLD) images and ERP studies showed a reduction in oxygenation and activity in the occipital visual cortex, insular cortex, cingulate cortex, precentral gyrus, hippocampus, and cerebellum in individuals staying at HA or HA natives [42].
Electroencephalogram (EEG) is used to evaluate the electrophysiological processing of cognitive activity, mostly after ascent to HA and in HA immigrants, but studies in HA natives are rare. In general, EEG shows that prolonged exposure to HA mainly impairs attention. In native adolescents in Bolivia, reductions in delta and beta frequency amplitude were recorded [43]. In Tibetan natives, a higher executive ability and impaired orienting ability were observed only at HA above 4000 m, suggesting a threshold for the influence of HA in brain function [7].
In highlanders living above 4000 m, lower levels of performance in executive functions are observed [8]. Researchers have also measured a reduction in spatial, abstract, and verbal abilities in adolescents living in El Alto, Bolivia (4150 m). HA significantly increases the risk for neurodevelopmental deficits, with larger effects on females [44]. On the contrary, attention deficit hyperactive disorder (ADHD) is less prevalent with increasing altitude, proposing a HA protective effect [45]. Thus, controversy is related to the possible neurocognitive function impairment at HA. However, the corresponding studies conducted so far have the drawback that they do not control biases associated with the socioeconomic level and family, school, and social environments.
In summary, highlanders have adapted to hypoxia through different physiological mechanisms (reviewed in [46]), such as increased lung capacity, increased ventilation, improved oxygen diffusion, increased total vessel density [47], and higher hemoglobin concentration. However, at very HA, impaired cerebral dynamic in blood flow [48], reduced cerebral glucose metabolism [49], reduced cerebral vascular reactivity [50][51], altered gray and white matter [42], and altered neuronal activity are some of the alterations that may affect neurocognitive functioning [52]. Understanding the molecular mechanisms underlying the brain adaptation to HA provides insights into developing interventions to mitigate the adverse effects of HA exposure, such as acute mountain sickness (AMS) and HA cerebral edema (HACE).
2. Hypoxia-Inducible Factors (HIFs) in the Brain Adaptation to HA
At HA, low oxygen supply leads to changes in gene expression mediated by the hypoxia-inducible factors (HIFs) pathway, which play a key role in the cellular response to hypoxia. In 2019, the Nobel Prize in Physiology was awarded to William Kaelin, Peter Ratcliffe, and Gregg Semenza for the discovery of HIFs as master regulators of oxygen homeostasis [53]. HIFs are heterodimers composed of an oxygen-sensitive alpha-subunit (HIF-α) and a constitutive beta-subunit (HIF-1β, also known as ARNT, aryl hydrocarbon receptor nuclear translocator). The two major HIF-α isoforms are HIF-1α and HIF-2α. Although the kinetics of stabilization and transactivation of both isoforms are similar [54], recent genome-wide studies have identified that the EPAS1 gene that encodes for HIF-2α is the major isoform involved in HA adaptation [55]. EPAS1 distribution is tissue- and cell-specific, mainly expressed in organs such as the kidney [56][57] and brain [58].
Hypoxic HIF-2α stabilization at HA leads to the formation of the HIF-2 complex that activates the expression of erythropoietin (EPO). EPO is a glycoprotein hormone that, when released from the kidney, enhances the formation of red blood cells (erythropoiesis) in the bone marrow to increase the blood’s oxygen-carrying capacity [59]. In the brain, EPO regulates the neural respiratory zones (central and peripheral), leading to higher HVR, thereby increasing tissue oxygenation [26]. EPO also increases brain angiogenesis (formation of new vessels) [3][60], neurogenesis (formation of new neurons) [3], synaptogenesis (new synapses) [61], and brain oxidative metabolism and cognition [62][63] (Figure 2).
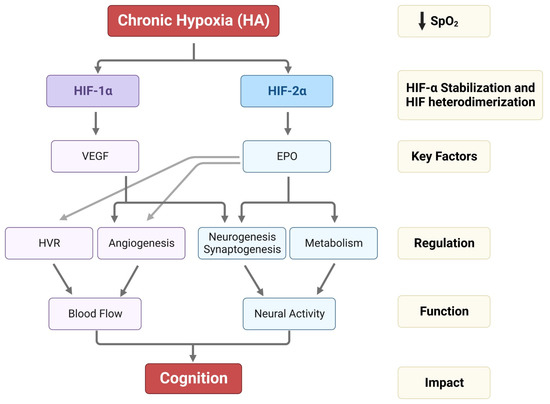
Figure 2. Impact of HA on HIF-1α, HIF-2α stabilization, VEGF and EPO expression, and their regulation in HVR, angiogenesis, neurogenesis, synaptogenesis, and metabolism. The alterations in HVR, angio-neurogenesis, and metabolism influence brain function and cognition.
On the other hand, the HIF-1 complex controls the expression of vascular endothelial growth factor (VEGF), an important signaling protein involved in vasculogenesis, angiogenesis (reviewed in [64]), and neurogenesis [65]. After 11 days of ascent to a HA, researchers observed that inhibition of VEGF signaling in rats interferes with vasculogenesis, angiogenesis, and neurogenesis. In contrast, EPO mainly involves HA-mediated angiogenesis in non-neurogenic brain areas [3] (Figure 2). In Sherpa highlanders, where tolerance to hypoxia is partly attributed to an increase in microcirculatory blood flow and capillary density, the VEGFA plasma levels did not correspond to HA and remained equivalent to the level in non-Sherpa low landers. This finding was speculated to be associated with distinctive genetic variations in the promoter region of VEGFA [66]. Conversely, an increase in VEGFA and EPO in the Andean population is related to chronic mountain sickness (CMS) [67].
HIF-1 also controls the expression of brain-derived neurotrophic factor (BDNF) and insulin-like growth factor-I (IGF-I), both key growth factors that regulate neurogenesis and synaptogenesis from embryonic to adult stages (reviewed in [68][69]). BDNF also interacts with astrocytes and neurons to control respiration [70][71][72]. Animal and human research indicate that physical activity can enhance BDNF blood levels and gene expression [73]. In contrast, long-term exposure to hypoxia lowers BDNF serum levels [74], yet the impact of HA on BDNF expression remains unexplored. No changes in IGF-I expression were observed after acute or chronic exposure to HA [75].
HIF-1 and -2 are crucial for proper brain development and continuously expressed in adult brain neurogenic zones and neural stem/progenitor cells from the embryonic and postnatal mouse brain [76]. HIF-1α deficient mice exhibit hydrocephalus accompanied by reduced neuronal cells and spatial memory impairment [77]. Additionally, HIF-2 has been shown to protect neural progenitor cells and neural differentiation processes by upregulating the survival orthologues Birc5a and Birg5b during embryogenesis [78]. Moreover, recent studies showed that loss of HIF-2α can affect cognitive performance in mice and causes a loss of pyramidal neurons in the retrosplenial cortex, a brain area responsible for spatial navigation [58]. This specific role of HIF-2 in neural development and synaptic plasticity suggests that it is crucial for cognitive function. Thus, targeting HIF-mediated modifications may hold therapeutic potential for HA-related illnesses such as AMS, HACE, and CMS neurologic deficits.
3. Signaling Pathways Interacting with HIFs
Neuronal viability is maintained through a complex interacting network of signaling pathways that can be disturbed in response to many cellular stresses. An imbalance in these signaling pathways after stress or in response to pathology can have drastic consequences for the function or fate of neurons. The mechanisms underlying the HIF-mediated changes in the brain involve a complex interplay between various signaling pathways, including the cyclic adenosine monophosphate (cAMP) pathway, phosphoinositide 3-kinase (PI3K) pathway, and nitric oxide (NO) pathway.
The cAMP pathway is an important second messenger system that plays a critical role in regulating numerous neural processes in the brain, from development, cellular excitability, synaptic plasticity, learning, and memory (reviewed in [79][80][81]). HIF-1 activates the cAMP pathway in response to hypoxia in cancer cells [82][83]. In the brain, cAMP leads to increased expression of the N-methyl-D-aspartate (NMDA) receptor subunit GluN1 in the hippocampus, an area involved in learning and memory [84]. cAMP can also regulate the activity of various downstream effectors involved in synaptic plasticity and memory formation, such as the protein kinase A (PKA) [85] and the cAMP response element-binding protein (CREB) [86]. Moreover, the cAMP signaling pathway can modulate other molecular targets in the brain, such as mitochondrial function and oxidative stress, by activating CREB and the peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) [87]. A longitudinal cohort study of proteomic and clinical biomarkers of AMS symptom phenotypes in 53 individuals after ascent to HA showed that the cAMP pathway interferes with the symptoms of AMS [88]. Therefore, this pathway could be a potential therapeutic strategy for AMS and understanding acclimatization to HA.
The Wnt/β-catenin pathway is directly regulated by HIF and is involved in developing brain structures such as the cerebral cortex and hippocampus (reviewed in [89]). The key mediator of Wnt signaling, the armadillo protein-β-catenin, participates in transcriptional regulation and chromatin interactions and is regulated by hypoxia [89]. The Wnt-mediated activation of HIF-1 promotes the maintenance of embryonic and neural stem cell activity and is significantly decreased in differentiated cells [90]. Furthermore, chronic exposure to hypoxia in vivo induces activation of the Wnt/β-catenin signaling cascade in the hippocampus, suggesting that mild hypoxia may have therapeutic value in neurodegenerative disorders [91].
The PI3K/Akt pathway is another important signaling pathway critical in regulating cell growth and survival and has been widely reported in brain development, aging, neurodegenerative diseases, and psychotic disorders (reviewed in [92]). HIF-1 and PI3K/Akt may interact on functional and regulatory levels. PI3K/Akt is required for heat shock proteins to protect HIF-1α from Von-Hippel-Lindau (pVHL)-independent degradation [93]. However, another earlier study has refuted this dependent interaction [94]. In a rat model of Alzheimer’s disease, it has been shown that gamma-aminobutyric acid (GABA) type B receptor-mediated PI3K/Akt activation alleviates oxidative stress and neuronal cell injury [95]. Moreover, in cerebral ischemic injury, the co-activation of GABAA and GABAB receptors exerted a neuroprotective effect via the PI-3K/Akt pathway [96]. Also, activation of the PI3K/Akt pathway has been shown to have neuroprotective effects and improve cerebral blood flow in animal models of cerebral ischemia [97]. Therefore, targeting this pathway could be a potential therapeutic strategy to treat or prevent HACE. One study demonstrated that treating hypoxic mouse brain microvascular endothelial cells with a PI3K activator, 3-methyladenine, suppressed hypoxia-induced endothelial permeabilization [98] and might effectively tackle HACE. Increased Akt activation (and overexpression of Parkin—a molecule that plays a critical role in ubiquitination) has been shown to reduce hypoxia-induced death of induced pluripotent stem cells-derived neurons from CMS. It is proposed that increased Akt activation protects against hypoxia-induced cell death. Therefore, it is suggested that impaired adaptive mechanisms, including lack of Akt activation and increased Parkin expression, render neurons from chronic mountain sickness subjects more susceptible to hypoxia-induced cell death [99].
The NO signaling pathway plays a significant role in HA-related illnesses, particularly in regulating vascular function and blood flow (reviewed in [100]). Hypoxia-induced HIF activation leads to increased expression of endothelial NO synthase (eNOS) [101]. NO acts as a vasodilator, causing the relaxation of vascular smooth muscle cells, increasing blood flow to oxygen-deprived tissues, and maintaining endothelial homeostasis [102]. NO also plays a pivotal role in regulating neurotransmitter release, such as acetylcholine, catecholamines, and neuroactive amino acids [103], as well as synaptic plasticity in the cerebral cortex [104]. Alterations in NO signaling have been implicated in several HA-related illnesses, including pulmonary edema and HACE [100]. In HA pulmonary edema, impaired NO production and increased pulmonary vasoconstriction can lead to increased fluid accumulation in the lungs. Tibetans have NO levels in the lung, plasma, and red blood cells that are at least double and, in some cases, orders of magnitude greater than in other populations, regardless of altitude [100]. Therapeutic interventions targeting the NO signaling pathway, e.g., administering NO donors or phosphodiesterase inhibitors, have shown promise in preventing and treating HA-related illnesses [105]. HIF-2-regulated EPO induced NO release in endothelial cells [106], cardiomyocytes [107], and lung cancer cells [108]. NO, in turn, co-regulated mitochondria and energy metabolism in combination with Akt. A similar mechanism was witnessed in the murine postnatal hippocampus, corresponding to enhanced cognition in early adulthood [63]. Moreover, NO (and derived S-nitrosothiols and glutathionylation) is required for the physiological response to hypoxia. NO directly interacts with brainstem respiratory centers and modulates the ventilatory response to hypoxia [109]. Furthermore, the current findings suggest that NO’s activity mediates the excitatory and inhibitory components of the hypoxic ventilatory response, and that NO may play a role in modulating the prominent second phase of the biphasic response to hypoxia [110].
Glutathionylation is crucial in redox signaling and cellular adaptation to oxidative stress. It is an important mechanism in regulating the activity of various proteins, including ion transporters and hypoxia-inducible factor 1 (HIF-1) pathway proteins. Sodium/Potassium ATPase (Na+/K+-ATPase) is an essential membrane protein responsible for maintaining the electrochemical gradient across the plasma membrane of cells. Hypoxia induces glutathionylation of specific cysteine residues in the α-subunit of Na+/K+-ATPase. This modification can alter the activity of the pump, affecting ion balance and cellular function [111][112]. Also, glutathionylation of sarcoplasmic/endoplasmic reticulum (ER) calcium ATPase (SERCA) occurs under hypoxia, leading to altered calcium handling and ER stress [113]. Glutathionylation can also affect the DNA-binding activity of HIF-1α. It has been suggested that glutathionylation of specific cysteine residues in HIF-1α may enhance its binding to the hypoxia response elements (HREs) present in the promoter regions of target genes, thereby influencing their transcriptional activation [114]. Glutathionylation of proteins, including ion transporters and HIF-1, is a crucial mechanism in the cellular response to hypoxia. It allows cells to modulate the activity of proteins involved in ion balance, calcium signaling, and gene expression, enabling adaptation and survival under conditions of oxygen limitation. Further research is needed to fully understand the specific cysteine residues targeted for glutathionylation and the functional consequences of this modification on protein activity in the context of hypoxia.