Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Victoria V. Shumyantseva | -- | 2435 | 2023-06-22 10:02:36 | | | |
| 2 | Lindsay Dong | Meta information modification | 2435 | 2023-06-25 03:22:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Shumyantseva, V.V.; Koroleva, P.I.; Bulko, T.V.; Agafonova, L.E. Alternative Electron Sources for Cytochrome P450s Catalytic Cycle. Encyclopedia. Available online: https://encyclopedia.pub/entry/45965 (accessed on 28 July 2026).
Shumyantseva VV, Koroleva PI, Bulko TV, Agafonova LE. Alternative Electron Sources for Cytochrome P450s Catalytic Cycle. Encyclopedia. Available at: https://encyclopedia.pub/entry/45965. Accessed July 28, 2026.
Shumyantseva, Victoria V., Polina I. Koroleva, Tatiana V. Bulko, Lyubov E. Agafonova. "Alternative Electron Sources for Cytochrome P450s Catalytic Cycle" Encyclopedia, https://encyclopedia.pub/entry/45965 (accessed July 28, 2026).
Shumyantseva, V.V., Koroleva, P.I., Bulko, T.V., & Agafonova, L.E. (2023, June 22). Alternative Electron Sources for Cytochrome P450s Catalytic Cycle. In Encyclopedia. https://encyclopedia.pub/entry/45965
Shumyantseva, Victoria V., et al. "Alternative Electron Sources for Cytochrome P450s Catalytic Cycle." Encyclopedia. Web. 22 June, 2023.
Copy Citation
The functional significance of cytochrome P450s (CYP) enzymes is their ability to catalyze the biotransformation of xenobiotics and endogenous compounds. P450 enzymes catalyze regio- and stereoselective oxidations of C-C and C-H bonds in the presence of oxygen as a cosubstrate. Initiation of cytochrome P450 catalytic cycle needs an electron donor (NADPH, NADH cofactor) in nature or alternative artificial electron donors such as electrodes, peroxides, photo reduction, and construction of enzymatic “galvanic couple”.
cytochrome P450
photocatalysis
biocatalysis
electron transfer
electrochemistry
1. Introduction
Cytochrome P450s (CYPs) are unique enzymes with great transforming and synthetic activities. CYPs possess unique catalytic activities such as monooxygenase, oxygen reductase or substrate reductase. CYPs participate in chemical reactions of heteroatom oxygenation and dealkylation, aromatic and aliphatic hydroxylation, and cleavage of esters and oxidation of double bonds [1][2][3]. The P450 monooxygenase system is capable of metabolizing medicinal drugs, pollutants, carcinogens, and steroid hormones. Cytochrome P450 monooxygenases exhibit great potential for application in the role of bioreactors for the decomposition of a variety of hydrophobic chemicals, including pollutants. CYPs also possess stereo specificity for the biosynthetic application and synthesis of new drugs or metabolites with a new spectrum of pharmacological activities [4][5]. The development of new innovative approaches based on the functional role of CYPs is a prospective and promising method for the practical use of these enzymes in toxicology, pharmacology, new drug metabolism, and biomedical application. However, CYPs as hemoproteins cannot implement such a great variety of biotransformation reactions alone and need protein redox partners. Type I P450 catalytic system is a three-component one, in which the redox partners represent a FAD-containing ferredoxin reductase and iron-sulfur ([Fe-S]) protein ferredoxin. The mitochondrial and bacterial CYP systems belong to a Type I system. Type II P450 catalytic system is a two-component metabolic machine that consists of FAD/FMN-containing flavoprotein, NADPH-dependent cytochrome P450-reductase (CPR), and transfers the reducing equivalents to hemoprotein. Microsomal CYP systems are Type II (class II). Type III is a self-sufficient one-component system with a reductase and heme domain on one polypeptide chain [6][7][8][9][10].
2. Active Metals as Electron Donor for the Reduction in Heme Iron of CYPs
The application of active metals as alternative electron sources is a prospective approach in the construction of effective biocatalytic systems. Additional equipment is not necessary for active metal application. The system only needs the appropriate protein and active metal to supply the system with electrons. The starting step in the CYP electron transfer chain is the reaction of ferric (Fe3+) with one electron leading to the formation of ferrous (Fe2+) [4][9][10]. The operating principle of a galvanic element is based on the difference in the electrode potentials of the oxidant and the reducing agent. For (Fe3+) +1 electron → (Fe2+) reaction reduction potential is +0.77 V (vs. SHE, standard hydrogen electrode) [11]. Active metals (e.g., Zn, Mg, Ti) are the most commonly used as reducing agents, while hemoproteins are used as oxidants. Hemoproteins have a more positive standard electrode potential than zinc, such as E0 (Zn0/Zn2+) = −0.763 V; therefore, zinc serves as a reducing agent in a zinc-hemoprotein “biogalvanic element” (the NADPH/NADP+ cofactor redox potential is −0.32 V (vs. SHE) in aqueous media). Scheme 1 represents a relative range of metal activity based on redox potentials vs. SHE.
Scheme 1. A relative range of metal activity vs. standard hydrogen electrode.
For modelling CYP-catalyzed reactions, an artificial hemoprotein was designed. Upon the complex formation of human serum albumin with iron protoporphyrine IX, there occurred the incorporation of heme into the protein and the formation of a specific complex with the albumin to heme molar ratio 2:1.
Cytochrome P450BM3 (CYP102A1) is Type III self-sufficient flavohemoprotein with heme and flavin domain on one polypeptide chain [2][3][12]. Cytochrome P450BM3 (CYP102) is a water-soluble, NADPH-dependent fatty acid hydroxylase from Bacillus megaterium that catalyzes the sub-terminal oxidation of saturated and unsaturated fatty acids. Several mutants of cytochrome P450BM3, such as P450BM3 M7 mutant (F87A V281G M354S R471C A1011T S1016G Q1022R) and P450BM3 M9 (R47F F87A M238K V281G M354S D363H W575C A595T) with improved electron transfer rate have been obtained [12]. P450BM3 M7 and P450BM3 M9 mutants were immobilized on DEAE-650S, further entrapped with k-carrageenan together with zinc dust, which functions as an electron source. For the effective reduction in CYP102 mutants, Zn/cobalt sepulchrate3+ complex (Zn/Co(III)sep) was used. Zn served as electron donor, Co(III)sep (S)-[1,3,6,8,10,13,16,19-octaazabicyclo-[6,6,6,]eicosane)cobalt(III)3+) served as electron transfer mediator.
3. Light-Driven CYP Catalysis
The aim of the elaboration of CYP-based systems with alternative electro sources is to avoid expensive NADPH cofactor and protein redox partners [2][3][7]. For the effective photochemical electron transfer, three main compounds should be present for the realization of the light-dependent P450 catalysis (Figure 1).

Figure 1. Schematic representation of the light-driven heme iron reduction. R-substrate, ROH-metabolite of CYP-dependent reaction.
Light sources → Sacrificial electron donors → Photosensitizer → Heme iron
Different types of photosensitizers have been used for the light-driven reduction of CYP enzymes, such as biological Photosystem I, nanomaterials, quantum dots (CdS, TiO2, and Ag nanoparticles), prosthetic group analogs (deazaflavins, isoalloxazine-type flavins), organic dyes (eosin Y), inorganic metal complexes, such as ruthenium polypyridine ([Ru(bpy)3]2+), zinc porphyrin, and their derivatives [13][14].
The electron transfer cascade in the photosensitizers/protein complex may be realized as a covalent or non-covalent binding, permitting the variation of the mode of interaction [1][15][16].
Sacrificial electron donors (sacrificial reductants) include ascorbic acid, triethanolamine (TEOA), diethyldithiocarbamate (DTC), 2-(N-morpholino), ethanesulfonic acid (MES), and ethylenediaminetetraacetic acid (EDTA) [17].
For effective irradiation, light sources with different power and light emitting spectrum in the visible region are used as a source of energy, and varied distances from the reaction cell must also be the subject of discussion [12][13][14][15][17][18][19][20].
Reductase cofactors, such as flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), or riboflavin, can be reduced photochemically using ethylenediaminetetraacetic acid (EDTA) as an electron donor [12]. During photo activation, reduced flavins can generate hydrogen peroxide, so peroxide shunt pathway is realized [18].
Bacterial cytochromes P450BM3 (CYP CYP102), CYP199A4, from Rhodopseudomonas palustris HaA2 (as T252E mutant), algal P450s, artificial hemoprotein as a complex of human serum albumin and heme, reductase-free human CYPs expressed in Escherichia coli were used in photobiocatalytic systems [15][16][20][21][22][23][24].
Whole cells expressing in Escherichia coli human CYPs 1A1, 1A2, 1B1, 2E1, and 3A4 for the bioconversion of marketed drugs and steroids were conducted to demonstrate the general applicability of the photobiocatalytic system [23][25]. Substrate conversion was registered for 4-nitrophenol as model substrate and drugs chlorzoxazone, lovastatin, simvastatin, and 17β-estradiol. For whole-cell photocatalysis, the authors observed the transport of flavins into the Escherichia coli cells producing CYP2E1 by cytometric analysis based on fluorescent intensity assay.
4. Electrochemical Technology for Effective CYP Catalysis
Despite the great potential of cytochrome P450s, the dependence on expensive nicotinamide cofactor (NADPH) and protein redox partners, such as NADPH-P450 reductase (CPR), limits their employment in synthetic chemistry, pharmacology, nanobiotechnology and industry [1][2][3][7][8][26]. Since the catalytic cycle of cytochromes P450 is associated with the transfer of electrons [1][2][3], the use of electrochemical systems has found its practical application for modeling catalytic reactions of this class of hemoproteins. In electrochemical systems, electrodes can supply enzymes with electrons instead of NADPHs. For efficient electron transfer, the modification of electrodes and immobilization of enzymes on electrodes are necessary [27][28][29][30]. Electroanalytical methods demonstrate privileges such as high analytical sensitivity, the application of disposable or reusable electrodes that can be modified with a broad spectrum of nanocomposite materials to obtain smart electrodes, the development of both analytical and compact equipment with friendly software for the registration and analysis of the data obtained. Bioelectronics initiation of cytochrome P450s catalysis needs an appropriate type of electrode with a rational design of sensor modifiers [27]. Electrochemical methods are a modern, highly sensitive analytical tool for studying various functional aspects of cytochromes P450: the search for substrates, inhibitors, effectors, and activators as new potential drug candidates [28][29][30][31][32][33][34][35].
4.1. Modelling on Electrode the Catalytic Cycle of CYP3A4
The biocatalytic mechanism of cytochrome P450s is very intricate and consists of several steps with intermediates formation [1][2][7]. The classical and well-known mechanism of CYP is represented in (Figure 2).
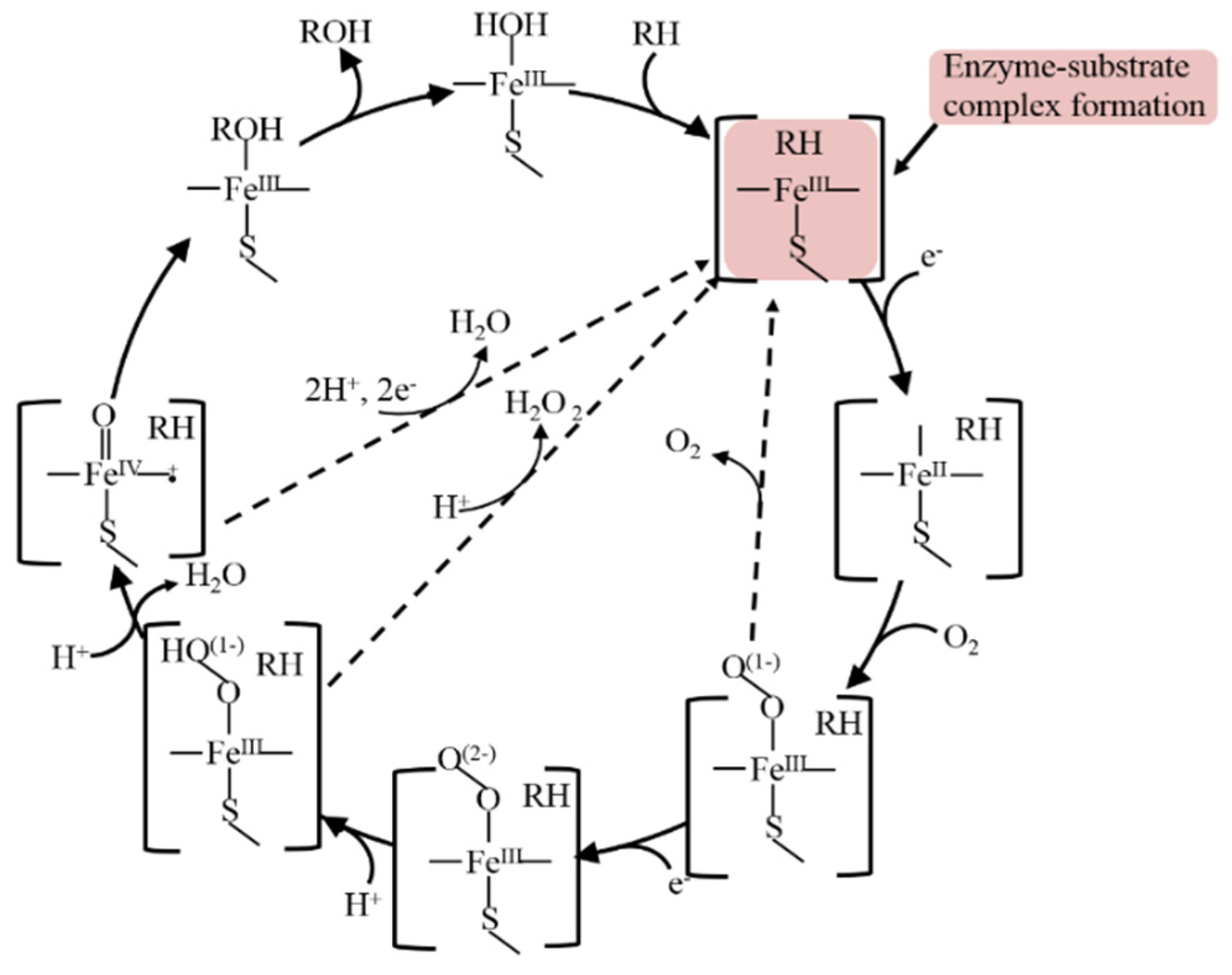
Figure 2. A common catalytic cycle of CYPs enzymes.
Substrate (RH) binds to enzyme (E) with productive enzyme-substrate complex formation at the first step of CYP catalysis, with productive complex formation inducing a spin shift, which allows an Fe(III)-to-Fe(II) reduction [1][2][7][33][34][35]. The confirmation of the formation of an enzyme/substrate complex was first reaffirmed spectroscopically for the interaction of substrate 17-α-progesterone with an adrenal cortex microsomal enzyme [35].
4.2. Electron Transfer Chain Optimization on CYP-Electrode
The electrochemical cytochrome P450 reactions have great potential for specific drug sensing, for searching for new drugs, and as bioreactors with broad synthetic applications. For the construction of a CYP-based electrochemical reactor, genetically engineered microsomes, human liver microsomes (HLM), and rat liver microsomes were used as the main participants of the electrochemical cell [36][37]. Microsomes consist of all proteins, which provide effective electron transfer to the heme iron ion for initiating the catalysis, such as cytochrome P450, and their redox partner protein CYP-NADPH reductase (CPR). Different types of electrodes were used for the microsomes’ utilization as bioreactors, such as a gold electrode, carbon electrode (polished basal plane pyrolytic graphite (BPG), edge plane pyrolytic graphite (EPG), glassy carbon (GC), or high-purity graphite (HPG) electrodes and modified with multiwalled carbon nanotubes EPG [36][37].
Rat liver microsomes (RLMs) were used for the detection of aflatoxin B1 (AFB1) metabolites, participating in carcinogenesis [38]. Electrochemical rat liver microsome-based biosensor using a composite of gold nanoparticles adsorbed on MXene (Au@MXene) for the rapid screening of AFB1. MXene is a new two-dimensional layered material-MXene which consists of transition metal carbides, nitrides, and carbonitrides. Rat liver microsomes (RLMs) were adsorbed on the Au@MXene nanocomposite and used for the detection of aflatoxin M1 in biosensor mode with a limit of detection of 2.8 nM.
In spite of the effective substrate conversion, liver microsomes as bioreactors or biosensors possess ethical problems dealing with the liver as microsomes’ source. Fro this reason, artificial systems modeling the electron transfer chains of CYP-dependent microsomes were proposed [39].
Efficient work of mitochondrial and microsomal cytochrome P450 systems requires additional redox proteins (diflavin reductase and cytochrome b5). NADPH-dependent cytochrome P450 reductase contains both FAD and FMN as prosthetic molecules and belongs to the flavoproteins group [1][7][8][9]. The roles of flavin nucleotides are the coupling of the reaction of hydroxylation of substrates, an increase in the efficiency of enzyme catalysis, regulation of the flow of electrons, as well as stimulation of positive conformational changes in the structure of the protein [8][40].
The G. Gilardi group proposed the construction of effective electron transfer chains using the “Lego” approach, combining the heme domain of bacterial CYP102 A1 (BM3), CYP116B5 or CYP3A4 and the reductase domain of BM3 [41][42][43][44][45][46][47][48][49]. These constructs demonstrated enhanced efficiency in electrochemical systems. It was shown that interprotein electron transfer occurred from reduced flavin(s) to heme iron in flavohemeproteins [50].
Riboflavin was used as the model of reductase for the optimization and simplification of the electron transfer chain. In the presence of riboflavin as a mediator of electron flow and NADPH as an electron donor, bacterial types of cytochromes P450 CYP106A2, CYP107DY1, CYP107DY1, HmtS, HmtT, HmtN efficiently catalyzed the reaction of N-dealkylation of substrate diphenhydramine [51].
The efficiency of catalysis of covalent and non-covalent complexes of riboflavin as a simulator of flavoprotein and cytochrome P450 2B4 from rabbit liver in the presence of NADH was investigated earlier [52][53]. Based on these experiments, scholars used riboflavin, FMN and FAD as substitutes for reductase flavoprotein for the enhancement/improvement of electron transfer in electrochemical cytochrome P450 systems (Scheme 2).
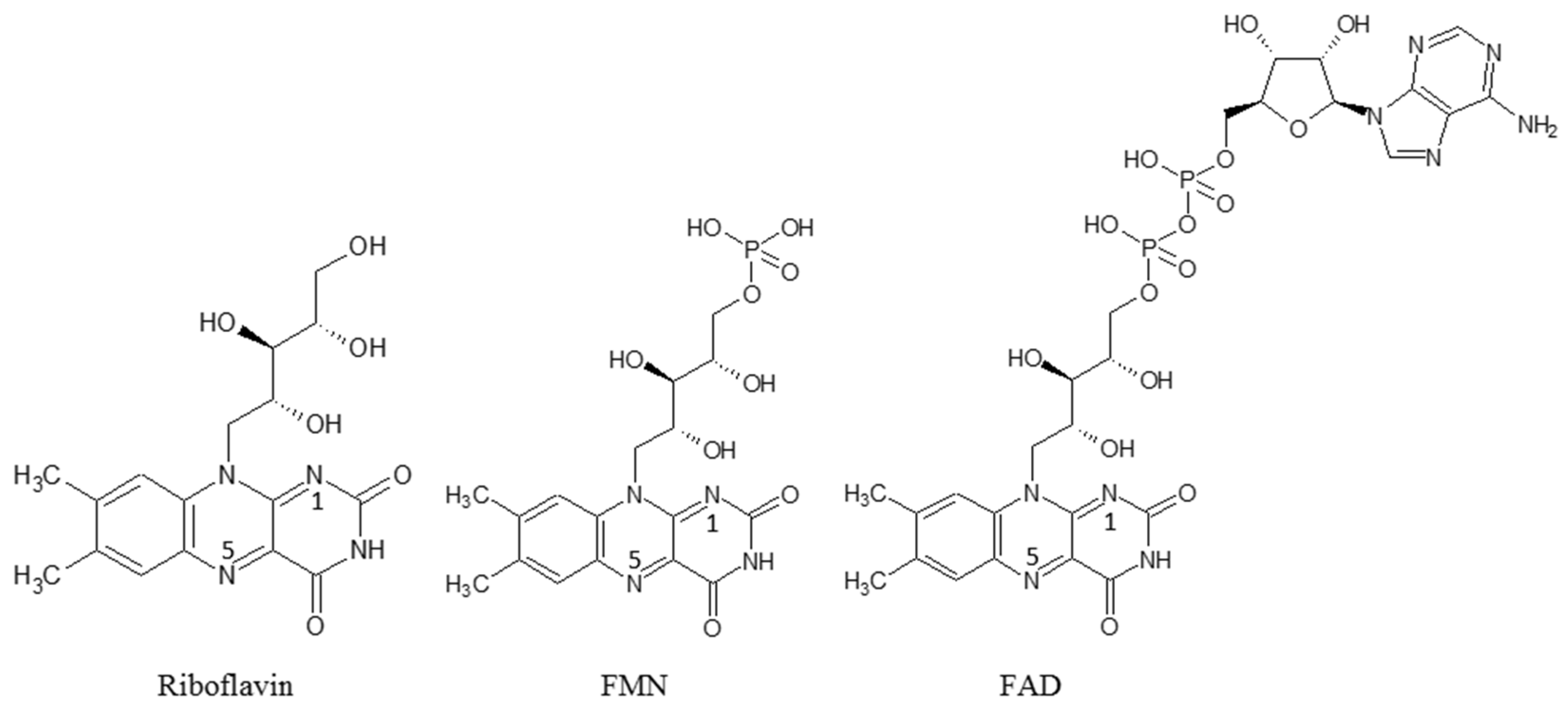
Scheme 2. Structures of flavin cofactors.
Hepatic enzyme cytochrome P450 3A4 (CYP3A4) is involved in the metabolism of about 50% of medicinal preparations and commercial drugs such as exogenous compounds. CYP3A4 catalyzes the metabolism of macrolide antibiotics (erythromycin, clarithromycin, azithromycin), calcium channel blockers (amlodipine, etc.), HIV protease inhibitors (indinavir, etc.), statins (such as simvastatin, atorvastatin), 5α-reductase inhibitors (finasteride), immunosuppressants (cyclosporine, etc.), antihistamines (astemizole), and prokinetics (cisapride) [5][6]. CYP3A4 also catalyzes the oxidation reactions of endogenous compounds, including estradiol (2- or 4-hydroxylation), testosterone (6β-hydroxylation), cortisol (6β-hydroxylation), cholesterol (4β-hydroxylation), progesterone (21-hydroxylation), cholic acid and chenodeoxycholic (formation of 3-dehydrocholic acid), and chenodeoxycholic (6α-hydroxylation) [5][6].
4.3. Modification of Electrode Surface for the 2D → 3D Transition
The efficient electron transfer from electrodes to biomolecules requires the immobilization of the protein on the working surface of the electrode. It is well-known that there is a problem with the interaction of proteins or enzymes with flat and “hard” 2D surfaces [54][55], which can lead to deformation, distortion of the protein’s spatial structure, or denaturation of the enzyme, which does not always have a positive effect on enzymatic/catalytic activity of immobilized proteins. When working with solid electrodes, modification of the working surface (for example, by polyion films, membrane-like compounds, self-assembled monolayers, and nanomaterials) not only promotes more efficient electron transportation but can lead to the stabilization of the tertiary structure of the protein [56]. Interaction with substrates also stabilizes the enzyme and can lead to changes in both electrochemical thermodynamics and parameters [54][56]. Enzymatic reactions in confined environments mimic the membranous surroundings and partially crowded cell media [57][58][59][60][61][62][63]. The 2D → 3D transition by means of the incorporation of enzymes into 3D nanopores on a plane electrode makes it possible to study the electrochemical and catalytic activity of enzymes [60][61][62][63].
The pore-forming protein streptolysin O (SLO) was proposed to mimic the cellular environment for creating a more developed surface with protein cavities for effective immobilization of CYP3A4 enzyme based on hybrid biomembranes in the lipid-like bilayer of the electrode modifier didodecyldimethylammonium bromide (DDAB) [62]. The novelty of the approach proposed is the enzyme incorporation in the three-dimensional composite DDAB/SLO, leading both to an improvement in the electron transfer properties and to the efficiency of CYP3A4 electrocatalysis. The confining effect when CYP3A4 is assembled inside cavities was investigated with direct non-catalytic voltammetry and electroanalysis of the enzymatic reaction, such as N-demethylation of erythromycin, occurring in the DDAB/SLO composite. SLO on the surface of lipid-like DDAB forms a highly developed surface with cavities, which permit the confinement of the CYP3A4 enzyme for direct non-catalytic and catalytic electrochemistry. The immobilized CYP3A4 demonstrated a pair of redox peaks with a formal potential of −0.325 ± 0.024 V. Potential of E = −0.5 V was applied for substrate conversion of erythromycin registered as N-demethylation reaction. The efficiency of erythromycin electrochemical N-demethylation in SPE/DDAB/CYP3A4 and SPE/DDAB/SLO/CYP3A4 were equal to 100 ± 22% and 297 ± 7%, respectively. AFM analysis of the SPE/DDAB/SLO revealed a more developed surface with protein cavities for the effective immobilization and confinement of the CYP3A4 enzyme.
References
- Bernhardt, R. Cytochromes P450 as Versatile Biocatalysts. J. Biotechnol. 2006, 124, 128–145.
- Bernhardt, R.; Urlacher, V.B. Cytochromes P450 as Promising Catalysts for Biotechnological Application: Chances and Limitations. Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203.
- Urlacher, V.B.; Girhard, M. Cytochrome P450 Monooxygenases in Biotechnology and Synthetic Biology. Trends Biotechnol. 2019, 37, 882–897.
- Paine, M.J.; Scrutton, N.S.; Munro, A.W.; Gutierrez, A.; Roberts, G.C.; Wolf, C.R. Electron transfer partners of cytochrome P450. In Cytochrome P450-Structure, Mechanism, and Biochemistry; Springer: Boston, MA, USA, 2005; pp. 115–148.
- de Montellano, P.R.O. Cytochrome P450: Structure, Mechanism, and Biochemistry; Springer: New York, NY, USA, 2015.
- Rendic, S.; Guengerich, F.P. Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 2015, 28, 38–42.
- Mi, L.; Wang, Z.; Yang, W.; Huang, C.; Zhou, B.; Hu, Y.; Liu, S. Cytochromes P450 in biosensing and biosynthesis applications: Recent progress and future perspectives. Trends Anal. Chem. 2023, 158, 116791.
- van Liempda, S.M.; Kool, J.; Reinen, J.; Schenk, T.; Meermana, J.H.N.; Irth, H.; Vermeulen, N.P.E. Development and validation of a microsomal online cytochrome P450 bioreactor coupled to solid-phase extraction and reversed-phase liquid chromatography. J. Chromatogr. A 2005, 1075, 205–212.
- Guengerich, F.P. Intersection of the Roles of Cytochrome P450 Enzymes with Xenobiotic and Endogenous Substrates: Relevance to Toxicity and Drug Interactions. Chem. Res. Toxicol. 2017, 30, 2–12.
- Guengerich, F.P. 1.19—Drug Metabolism: Cytochrome P450. In Comprehensive Pharmacology; Elsevier: Amsterdam, The Netherlands, 2022; pp. 470–508.
- Brett, C.M.A.; Oliveira Brett, A.M. Electrochemistry: Principles, Methods, and Applications, 1st ed.; Oxford Science University Publications: Oxford, UK, 1993.
- Zhao, L.; Güven, G.; Li, Y.; Schwaneberg, U. First steps towards a Zn/Co(III)sep-driven P450 BM3 reactor. Appl. Microbiol. Biotechnol. 2011, 91, 989–999.
- Zheng, D.; Zhang, Y.; Liu, X.; Wang, J. Coupling natural systems with synthetic chemistry for light driven enzymatic biocatalysis. Photosynth. Res. 2020, 143, 221–231.
- Zilly, F.E.; Taglieber, A.; Schulz, F.; Hollmann, F.; Reetz, M.T. Deazaflavins as mediators in light-driven cytochrome P450 catalyzed hydroxylations. Chem. Commun. 2009, 46, 7152–7154.
- Lassen, L.M.; Nielsen, A.Z.; Olsen, C.E.; Bialek, W.; Jensen, K.; Møller, B.L.; Jensen, P.E. Anchoring a plant cytochrome P450 via PsaM to the thylakoids in Synechococcus sp. PCC 7002: Evidence for light-driven biosynthesis. PLoS ONE 2014, 9, 102184.
- Eidenschenk, C.; Cheruzel, L. Ru(II)-diimine complexes and cytochrome P450 working hand-in-hand. J. Inorg. Biochem. 2020, 213, 111254.
- Nielsen, A.Z. Redirecting Photosynthetic Reducing Power toward Bioactive Natural Product Synthesis. ACS Synth. Biol. 2013, 2, 308–315.
- Jensen, K.; Jensen, P.E.; Møller, B.L. Light-driven chemical synthesis. Trends Plant Sci. 2012, 17, 60–63.
- Le, T.-K.; Park, J.H.; Choi, D.S.; Lee, G.-Y.; Choi, W.S.; Jeong, K.J.; Park, C.B.; Yun, C.-H. Solar-driven biocatalytic C-hydroxylation through direct transfer of photoinduced electrons. Green Chem. 2019, 21, 515.
- Shalan, H.; Kato, M.; Cheruzel, L. Keeping the spotlight on cytochrome P450. BBA-Proteins Proteom. 2018, 1866, 80–87.
- Shumyantseva, V.V.; Bulko, T.V.; Archakov, A.I. Regulation of cytochrome P450 activity by physicochemical methods. Uspekhi Khimii 1999, 68, 967–975.
- Zheng, S.; Guo, J.; Cheng, F.; Gao, Z.; Du, L.; Meng, C.; Li, S.; Zhang, X. Cytochrome P450s in algae: Bioactive natural product biosynthesis and light-driven bioproduction. Acta Pharm. Sin. B 2022, 12, 2832–2844.
- Le, T.-K.; Kim, J.; Nguyen, N.A.; Nguyen, T.H.H.; Sun, E.-G.; Yee, S.-M.; Kang, H.-S.; Yeom, S.-J.; Park, C.B.; Yun, C.-H. Solar-Powered Whole-Cell P450 Catalytic Platform for C-Hydroxylation Reactions. ChemSusChem 2021, 14, 3054–3058.
- Lee, J.H.Z.; Podgorski, M.N.; Moir, M.; Gee, A.R.; Bell, S.G. Selective Oxidations Using a Cytochrome P450 Enzyme Variant Driven with Surrogate Oxygen Donors and Light. Chem. Eur. J. 2022, 28, 202201366.
- Shumyantseva, V.V.; Bulko, T.V.; Zimin, A.G.; Uvarov, V.Y.; Archakov, A.I. Design of an artificial hemoprotein based on human serum albumin. Biochem. Mol. Biol. Int. 1996, 39, 503.
- Winkler, M.; Geier, M.; Hanlon, S.P.; Nidetzky, B.; Glieder, A. Human Enzymes for Organic Synthesis. Angew. Chem. Int. Ed. 2018, 57, 13406–13423.
- Shumyantseva, V.V.; Agafonova, L.E.; Bulko, T.V.; Kuzikov, A.V.; Masamrekh, R.A.; Yuan, J.; Pergushov, D.V.; Sigolaeva, L.V. Electroanalysis of Biomolecules: Rational Selection of Sensor Construction. Biochem. (Mosc.) 2021, 86, 140–151.
- Masamrekh, R.A.; Kuzikov, A.V.; Haurychenka, Y.I.; Shcherbakov, K.A.; Veselovsky, A.V.; Filimonov, D.A.; Dmitriev, A.V.; Zavialova, M.G.; Gilep, A.A.; Shkel, T.V.; et al. In vitro interactions of abiraterone, erythromycin, and CYP3A4: Implications for drug-drug interactions. Fundam. Clinic. Pharmacol. 2020, 34, 120–130.
- Shumyantseva, V.V.; Bulko, T.V.; Kuzikov, A.V.; Masamrekh, R.A.; Konyakhina, A.Y.; Romanenko, I.; Max, J.B.; Köhler, M.; Gilep, A.A.; Usanov, S.A.; et al. All-electrochemical nanocomposite two-electrode setup for quantification of drugs and study of their electrocatalytical conversion by cytochromes P450. Electrochim. Acta 2020, 336, 135579.
- Shumyantseva, V.V.; Kuzikov, A.V.; Masamrekh, R.A.; Bulko, T.V.; Archakov, A.I. From electrochemistry to enzyme kinetics of cytochrome P450. Biosens. Bioelectron. 2018, 121, 192–204.
- Masamrekh, R.; Filippova, T.; Haurychenka, Y.; Shumyantseva, V.; Kuzikov, A.; Shcherbakov, K.; Veselovsky, A.; Strushkevich, N.; Shkel, T.; Gilep, A.; et al. Estimation of the inhibiting impact of abiraterone D4A metabolite on human steroid 21-monooxygenase (CYP21A2). Steroids 2020, 154, 108528.
- Kostin, V.A.; Zolottsev, V.A.; Kuzikov, A.V.; Masamrekh, R.A.; Shumyantseva, V.V.; Veselovsky, A.V.; Stulov, S.V.; Novikov, R.A.; Timofeev, V.P.; Misharin, A.Y. Oxazolinyl derivatives of -21-norpregnene differing in the structure of A and B rings. Facile synthesis and inhibition of CYP17A1 catalytic activity. Steroids 2016, 115, 114–122.
- Krishnan, S. Bioelectrodes for evaluating molecular therapeutic and toxicity properties. Curr. Opin. Electrochem. 2019, 19, 20–26.
- Di Nardo, G.; Gilardi, G. Natural Compounds as Pharmaceuticals: The Key Role of Cytochromes P450 Reactivity. Trends Biochem. Sci. 2020, 45, 511–525.
- Sakaki, T. Practical application of cytochrome P450. Biol. Pharm. Bull. 2012, 35, 844–849.
- Nerimetla, R.; Krishnan, S. Electrocatalysis by subcellular liver fractions bound to carbon nanostructures for stereoselective green drug metabolite synthesis. Chem. Commun. 2015, 51, 11681–11684.
- Walgama, C.; Nerimetla, R.; Materer, N.F.; Schildkraut, D.; Elman, J.F.; Krishnan, S. A Simple Construction of Electrochemical Liver Microsomal Bioreactor for Rapid Drug Metabolism and Inhibition Assays. Anal. Chem. 2015, 87, 4712–4718.
- Sun, X.; Sun, J.; Ye, Y.; Ji, J.; Sheng, L.; Yang, D.; Sun, X. Metabolic pathway-based self-assembled liver microsome electrochemical biosensor for rapid screening of aflatoxin B1. Bioelectrochemistry 2023, 151, 108378.
- Faulkner, K.M.; Shet, M.S.; Fisher, C.W.; Estabrook, R.W. Electrocatalytically driven w-hydroxylation of fatty acids using cytochrome P450 4A1. Proc. Natl. Acad. Sci. USA 1995, 92, 7705–7709.
- Shumyantseva, V.V.; Koroleva, P.I.; Bulko, T.V.; Shkel, T.V.; Gilep, A.A.; Veselovsky, A.V. Approaches for increasing the electrocatalitic efficiency of cytochrome P450 3A4. Bioelectrochemistry 2023, 149, 108277.
- Gilardi, G.; Meharenna, Y.T.; Tsotsou, G.E.; Sadeghi, S.J.; Fairhead, M.; Giannini, S. Molecular Lego: Design of molecular assemblies of P450 enzymes for nanobiotechnology. Biosens. Bioelectron. 2002, 17, 133–145.
- Rua, F.; Sadeghi, S.J.; Castrignanò, S.; Di Nardo, G.; Gilardi, G. Engineering Macaca fascicularis cytochrome P450 2C20 to reduce animal testing for new drugs. J. Inorg. Biochem. 2012, 117, 277–284.
- Dodhia, V.; Sassone, C.; Fantuzzi, A.; Di Nardo, G.; Sadeghi, S.J.; Gilardi, G. Modulating the coupling efficiency of human cytochrome P450 CYP3A4 at electrode surfaces through protein engineering. Electrochem. Commun. 2008, 10, 1744–1747.
- Degregorio, D.; Sadeghi, S.J.; Di Nardo, G.; Gilardi, G.; Solinas, S.P. Understanding uncoupling in the multiredox centre P450 3A4-BMR model system. J. Biol. Inorg. Chem. 2011, 16, 109–116.
- Castrignanò, S.; Di Nardo, G.; Sadeghi, S.J.; Gilardi, G. Influence of inter-domain dynamics and surrounding environment flexibility on the direct electrochemistry and electrocatalysis of selfsufficient cytochrome P450 3A4-BMR chimeras. J. Inorg. Biochem. 2018, 188, 9–17.
- Castrignanò, S.; D’Avino, S.; Di Nardo, G.; Catucci, G.; Sadeghi, S.J.; Gilardi, G. Modulation of the interaction between human P450 3A4 and B. megaterium reductase via engineered loops. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 116–125.
- Degregorio, D.; D’Avino, S.; Castrignanò, S.; Di Nardo, G.; Catucci, G.; Sadeghi, S.J.; Gilardi, G. Human Cytochrome P450 3A4 as a Biocatalyst: Effects of the Engineered Linker in Modulation of Coupling Efficiency in 3A4-BMR Chimeras. Front. Pharmacol. 2017, 8, 121.
- Fantuzzi, A.; Meharenna, Y.T.; Briscoe, P.B.; Sassone, C.; Borgia, B.; Gilardi, G. Improving catalytic properties of P450 BM3 haem domain electrodes by molecular Lego. Chem. Commun. 2006, 12, 1289–1291.
- Correddu, D.; Catucci, G.; Giuriato, D.; Di Nardo, G.; Ciaramella, A.; Gilardi, G. Catalytically self-sufficient CYP116B5: Domain switch for improved peroxygenase activity. Biotechnol. J. 2023, 18, 2200622.
- Shumyantseva, V.V.; Bulko, T.V.; Lisitsyna, V.B.; Urlacher, V.B.; Kuzikov, A.V.; Suprun, E.V.; Archakov, A.I. Electrochemical Measurement of Intraprotein and Interprotein Electron Transfer. Biophysics 2013, 58, 349–354.
- Zhang, C.; Lu, M.; Lin, L.; Huang, Z.; Zhang, R.; Wu, X.; Chen, Y. Riboflavin Is Directly Involved in N-Dealkylation Catalyzed by Bacterial Cytochrome P450 Monooxygenases. Chembiochem 2020, 21, 2297–2305.
- Shumyantseva, V.; Uvarov, V.; Byakova, O.; Archakov, A. Semisynthetic Flavocytochromes Based on Cytochrome P450 2B4: Reductase and Oxygenase Activities. Arch. Biochem. Biophys. 1998, 354, 133–138.
- Shumyantseva, V.V.; Bulko, T.V.; Bachmann, T.T.; Bilitewski, U.; Schmid, R.D.; Archakov, A.I. Electrochemical Reduction of Flavocytochromes 2B4 and 1A2 and Their Catalytic Activity. Arch Biochem. Biophys. 2000, 377, 43–48.
- Martínez-Negro, M.; Oberländer, J.; Simon, J.; Mailänder, V.; Morsbach, S.; Landfester, K. A new methodology combining QCM-D and proteomic profiling enables characterization of protein adsorption on 2D surfaces. J. Colloid Interface Sci. 2023, 630, 965–972.
- Guengerich, F.P. Drug Metabolism: Cytochrome P450. Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2021.
- Lu, J.; Shen, Y.; Liu, S. Enhanced light-driven catalytic performance of cytochrome P450 confined in macroporous silica. Chem. Commun. 2016, 52, 7703–7706.
- Kuzikov, A.; Filippova, T.; Masamrekh, R.; Shumyantseva, V. Electroanalysis of 4′-Hydroxydiclofenac for CYP2C9 Enzymatic Assay. Electrocatalysis 2022, 13, 630–640.
- Fang, X.; Qiao, L.; Yan, G.; Yang, P.; Liu, B. Multifunctional nanoreactor for comprehensive characterization of membrane proteins based on surface functionalized mesoporous foams. Anal. Chem. 2015, 87, 9360–9367.
- Dai, Q.; Yang, L.; Wang, Y.; Cao, X.; Yao, C.; Xu, X. Surface charge-controlled electron transfer and catalytic behavior of immobilized cytochrome P450 BM3 inside dendritic mesoporous silica nanoparticles. Anal. Bioanal. Chem. 2020, 412, 4703–4712.
- Küchler, A.; Yoshimoto, M.; Luginbühl, S.; Mavelli, F.; Walde, P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 2016, 11, 409–419.
- Shumyantseva, V.V.; Koroleva, P.I.; Gilep, A.A.; Napolskii, K.S.; Ivanov, Y.D.; Kanashenko, S.L.; Archakov, A.I. Increasing the Efficiency of Cytochrome P450 3A4 Electrocatalysis Using Electrode Modification with Spatially Ordered Anodic Aluminum Oxide-Based Nanostructures for Investigation of Metabolic Transformations of Drugs. Dokl. Biochem. Biophys. 2022, 506, 215–219.
- Koroleva, P.I.; Gilep, A.A.; Kraevskiy, S.V.; Tsybruk, T.V.; Shumyantseva, V.V. Improving the Efficiency of Electrocatalysis of Cytochrome P450 3A4 by Modifying the Electrode with Membrane Protein Streptolysin O for Studying the Metabolic Transformations of Drugs. Biosensors 2023, 13, 457.
- Lin, S.; Chen, Y.; Wang, Y.; Cai, Z.; Xiao, J.; Muhmood, T.; Hu, X. Three-Dimensional Ordered Porous Nanostructures for Lithium−Selenium Battery Cathodes That Confer Superior Energy-Storage Performance. ACS Appl. Mater. Interfaces 2021, 13, 9955–9964.
More
Information
Subjects:
Biochemistry & Molecular Biology; Biophysics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
656
Revisions:
2 times
(View History)
Update Date:
25 Jun 2023
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No