 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Norbert Hoffmann | -- | 3742 | 2023-06-20 19:30:36 | | | |
| 2 | Sirius Huang | + 13 word(s) | 3755 | 2023-06-25 03:12:16 | | | | |
| 3 | Sirius Huang | Meta information modification | 3755 | 2023-06-25 03:13:54 | | | | |
| 4 | Sirius Huang | Meta information modification | 3755 | 2023-06-27 04:01:59 | | |
Video Upload Options
Biomass and biomass-derived compounds have become an important alternative feedstock for chemical industry. They may replace fossil feedstocks such as mineral oil and related platform chemicals. These compounds may also be transformed conveniently into new innovative bioactive products, for example, for the medicinal or the agrochemical domain.
1. Introduction

Figure 1. Graphical overview.
2. Photocatalytic Transformations of Biopolymers
3. Carbohydrates
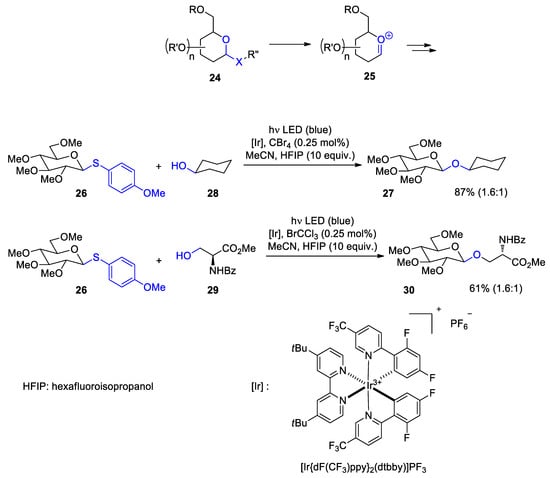
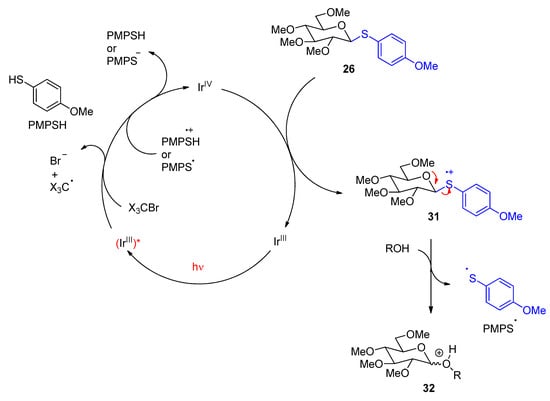
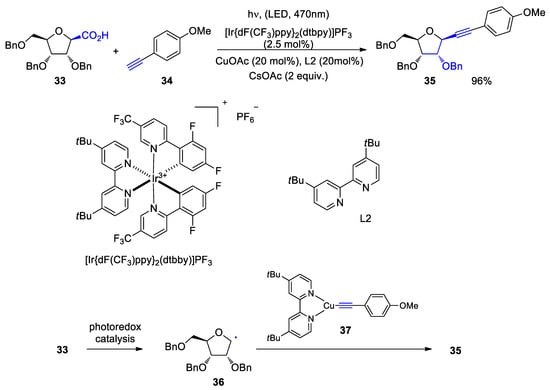
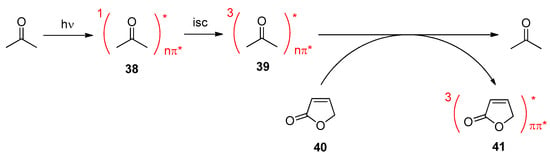
4. Fatty Acids
Fatty acids obtained from triglycerides play an important role in the chemical valorization of biomass. Their use for the production of surfactants belong to the basic culture techniques of humanity. More recently their application as part of biofuel is reported [142]. Fatty acid methyl esters (FAME) can be obtained by photocatalytic esterification to produce biodiesel [143][144][145].
Photodecarboxylation is an interesting transformation both for biofuel production and in organic synthesis [146][147][148] as it leads to radical intermediates that can form alkanes or further react with a variety of substrates. Decarboxylation is also carried out using photochemical electron transfer [149][150][151][152]. Such reaction steps also play an important role in many metal catalyzed reactions [130][149][153][154]. In this context, but also for application to organic synthesis, decarboxylation is an interesting transformation as it leads to a variety of radical intermediates.
Hatanaka et al. have transformed palmitic acid into the corresponding n-pentadecane [155]. This method consists of the photoelectron transfer of the carboxylate with a photogenerated cation radical of phenantrene followed by decarboxylation. The hydrogen abstraction from a thiol results in the formation of the corresponding alkane. Nicewicz et al. used of an acridinium photooxidant in the presence of an organic base for a decarboxylation [156]. The mechanism involves a photochemical electron transfer to the acridinium from the carboxylate followed by CO2 release. The generated carbon radical abstracts a hydrogen from the in situ generated thiophenol. Wang et al. reported a route for photocatalytic decarboxylation for alkane production using fatty acids. Their system uses semiconductor Pt/TiO2 and irradiation using 365 nm LEDs, and a hydrogen atmosphere resulted to be crucial for the high yields (>90%), while several fatty acids were successfully transformed in the corresponding alkanes [157].
Enzyme-catalyzed reactions play a key role in the transformation of biomass or biomass-derived compounds. Recently, an algal photoenzyme named fatty acid photodecarboxylase (CvFAP) was discovered. This photoenzyme transforms fatty acids into the corresponding alkanes upon light irradiation [158][159][160]. Among the different uses of this photo-enzyme, several methods were reported to obtain, for example, bio-propane and bio-butane from fatty acids [161][162], and also bio-diesel [163] depending on the substrate that was used.
Concerning the application of such reactions to organic synthesis, radical intermediates are often generated by decarboxylation of carboxylic acids or their derivatives [164][165][166][167][168]. These radicals undergo addition to various acceptor molecules. In many of these reactions, photocatalysis involving electron transfer is applied. The transformations are often also carried out with corresponding fatty acids or their derivatives.
5. Biomass-Derived Platform Chemicals
Levoglucosenone is an interesting platform chemical, which is obtained from pyrolysis of cellulose under acidic reaction conditions [169][170][171]. Recently, it gained additional interest as a precursor of a biobased dipolar solvent [172]. Levoglucosenone is now produced on the industrial scale. Different functional groups are localized on a relatively small enantiomerically pure molecule. Thus, it becomes a very attractive synthon for organic synthesis [173][174][175][176][177]. Levoglucosenone can also be transformed into further platform chemicals such as furanone derivatives [178][179].
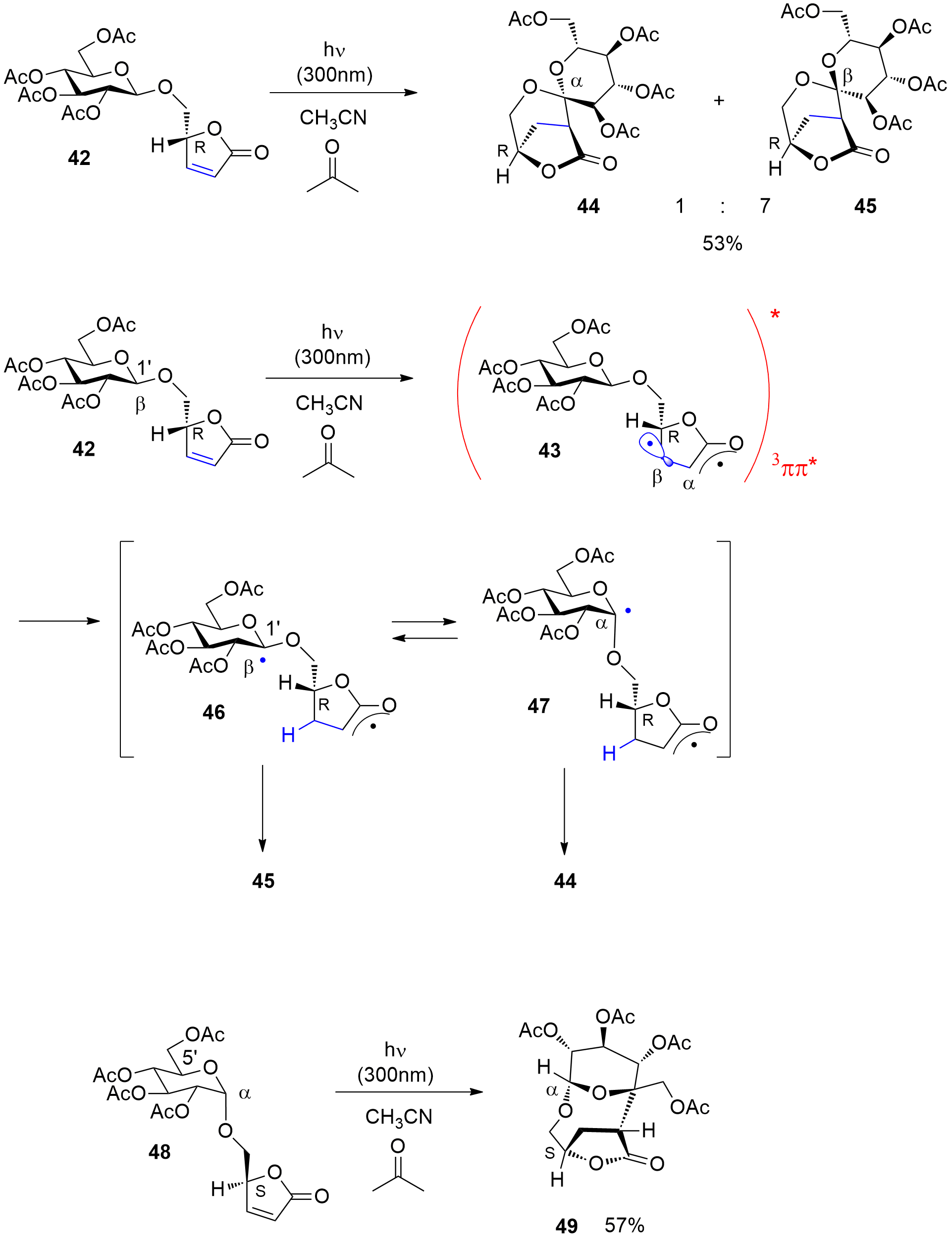
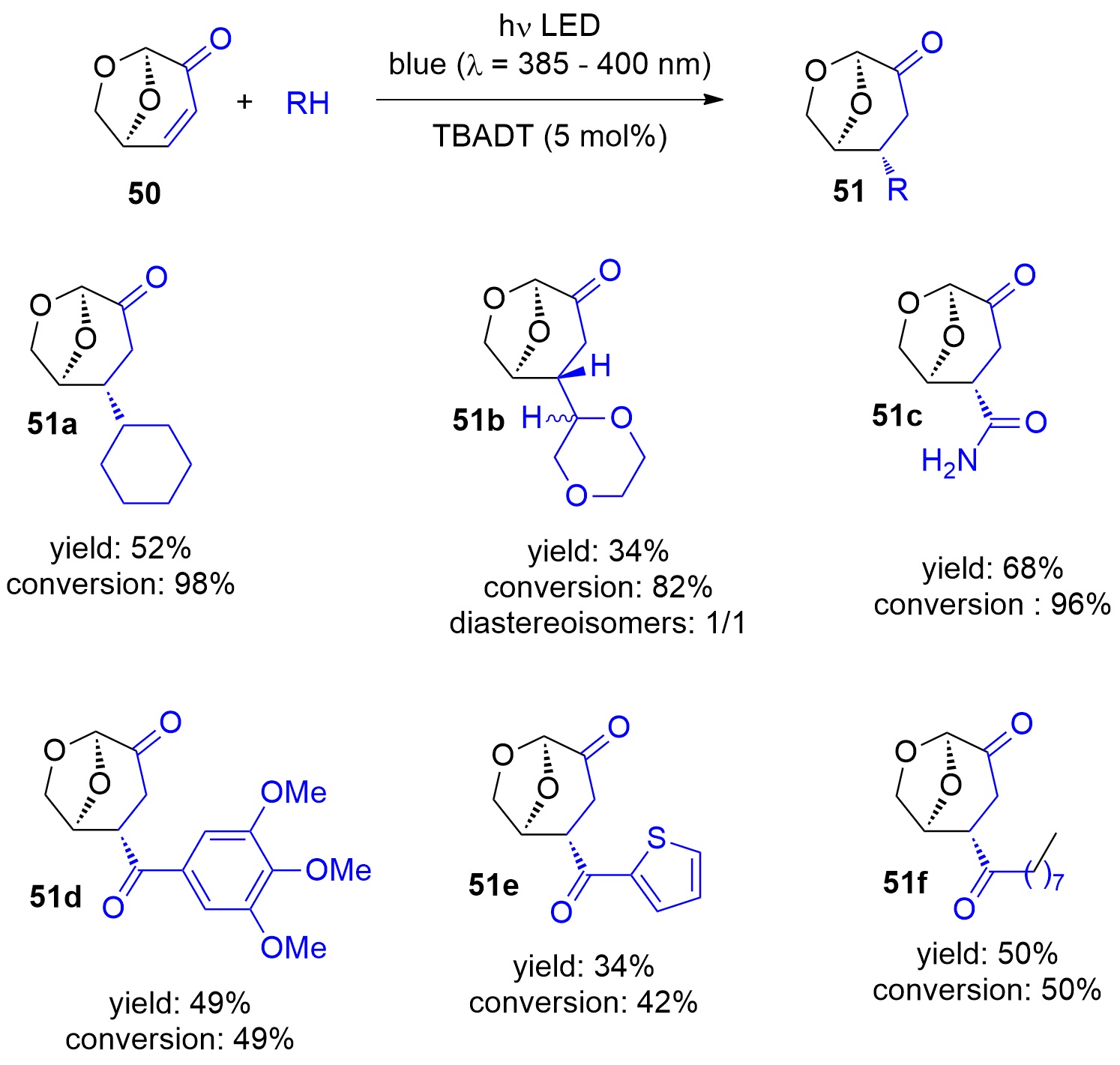
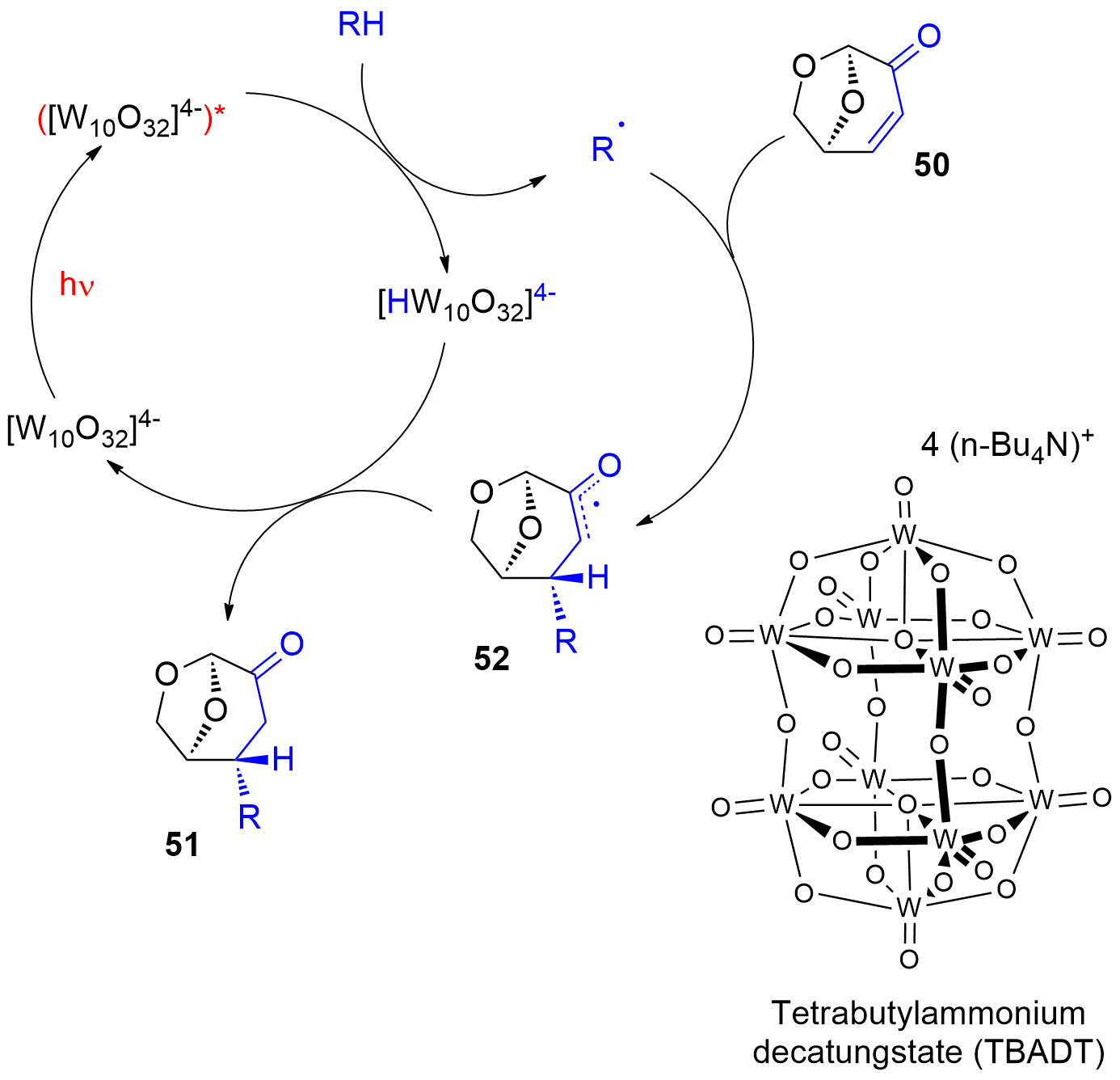
Photocatalytic reactions have also been performed with this compound. Using tetra-n-butylammonium decatungstate (TBADT) as a photocatalyst, various radical species have been added to levoglucosenone 50 (Scheme 10) [180]. Thus, alkanes (51a) or cyclic ethers (51b) have been added. The addition of formamide (51c) was particular efficient. The method was also used to the addition of aromatic (51d), heteroaromatic (51e) and aliphatic aldehydes (51f). The scope for the addition of aldehydes is large [181]. From these precursors radical intermediates are generated by hydrogen atom transfer (HAT) to the photochemically excited TBADT (Scheme 11) [182][183][184]. The radicals add stereospecifically anti with respect to the (‑CH2‑O‑) moiety in 50. The energy difference for both diastereotopic attacks of the radical intermediates is 5 kcal mol−1. The configuration of further chiral centers generated in the reaction as in 51b was not controlled. The electrophilic radical intermediate 52 is reduced by [HW10O32]4−, which leads to the final product 51. In the context of sustainable chemistry, it should further be mentioned that such reactions have been carried out with sunlight and under very simple apparatus conditions [183].
Scheme 10. Photocatalytic addition of radicals to levoglucosenone 50.
Scheme 11. Mechanism of the photocatalytic addition of radicals to levoglucosenone 50.
Furans are valuable platform chemicals obtained from carbohydrates [185][186][187]. They are formed by the dehydration of sugars. Thus, dehydration of pentoses or hemicellososes yields furfural while corresponding transformations of hexoses lead to hydroxymethylfurfural (HMF). These compounds are currently studied in connection with the production of bulk products such as new polymers [185][188] or surfactants [189][190][191].
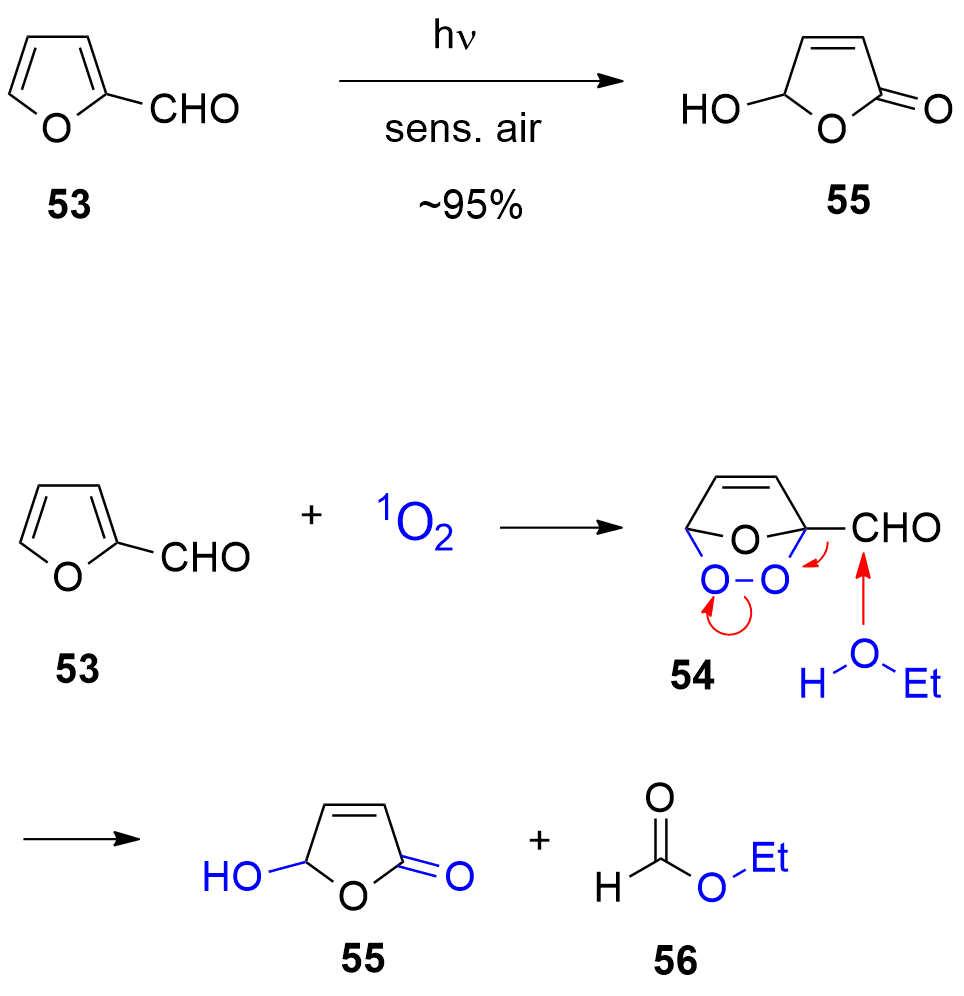
One of the most-used photocatalytic reactions of furans is the photooxygenation of these compounds involving singlet oxygen [192][193]. Oxygen is one of the rather rare molecules possessing a triplet multiplicity at the ground state [194][195]. The photooxygenation of furfural 53 with singlet oxygen yields hydroxyfuranone (Scheme 12) [196][197][198]. In most cases of photooxygenation of furan derivative, an endoperoxide such as 54 is formed. As the reaction is carried out in an alcohol as solvent, this intermediate undergoes a nucleophilic attack of an alcohol molecule leading to the final products 5-hydroxyfuran-2[5H]-one 55 and a formic ester 56. This very convenient oxidation can be carried out in the laboratory at 100 g scale [199]. Hydroxyfuranone 55 is itself a platform chemical with numerous applications to organic synthesis [200][201]. Chiral synthons can be obtained from this compound and many efficient applications to asymmetric synthesis have been reported [202][203][204][205]. Hydorxyfuranone 55 was also used for the preparation of biodegradable surfactants (Scheme 13) [206]. A photoredox catalytic process was also applied for the preparation of such compounds [207][208].
Scheme 12. Photooxygenation of furfural 53 to hydroxyfuranone 55.
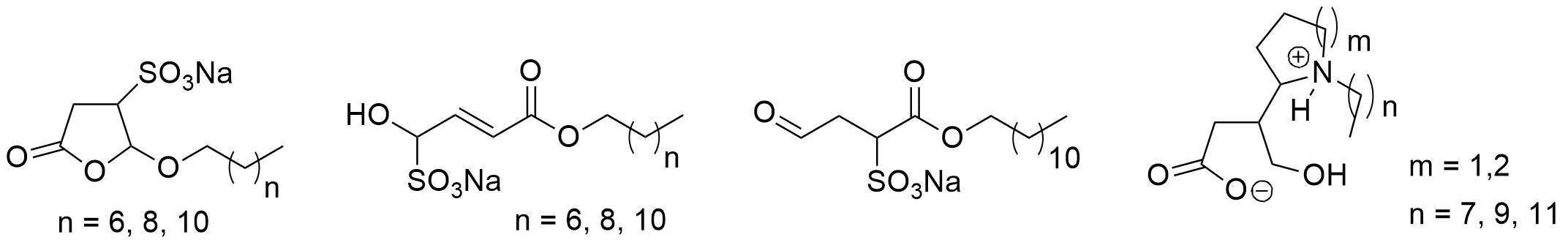
Scheme 13. Surfactants obtained from furfural.
Photooxygenation was also applied as a key step to the synthesis compound 57 (Scheme 14, also compare Scheme 9). Using conventional carbohydrate chemistry methodology, it is often difficult to prepare α-anomeric derivatives of hexose pyranoses such as derived from glucose for example. Isomaltulose is a disaccharide obtained by enzymatic isomerization of saccharose [209][210]. In the isomaltulose molecule, a fructose moiety in its furanose form is connected in the α-anomeric position of glucose. Selective dehydration on the fructose furanoside under acidic conditions leads to the formation a hydroxymethylfurfural derivative (58) [246]. Acylation (59) and photooxygenation yields the product 60. Finally, reduction of the hydroperoxide 60 leads to 57 [132]. The present synthesis also provides a very convenient access to 5-hydroxymethylfuranones such as 57. Using conventional methods of carbohydrate chemistry, these compounds are prepared with multi-step syntheses from sugars or amino acids [211][212].
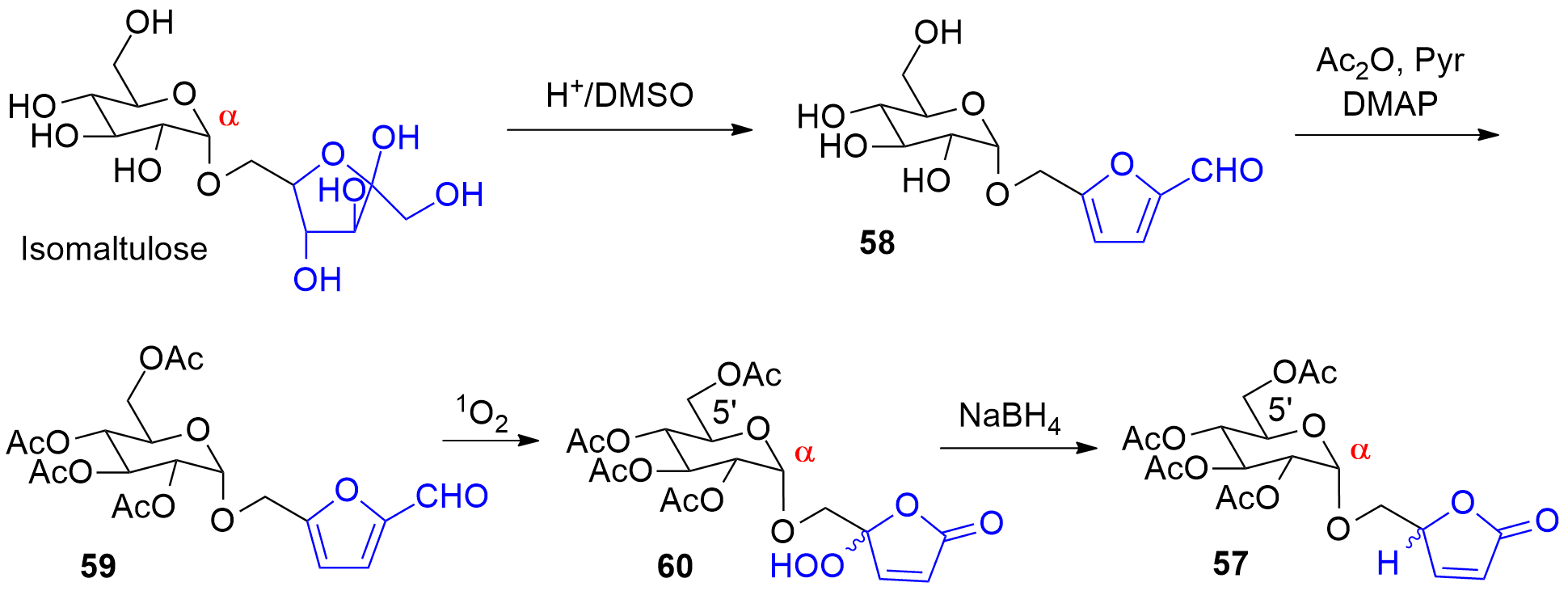
Scheme 14. Synthesis of a α-connected glycosyl furanone 57. from isomaltulose.
6. Conclusion
Biomass or biomass-derived platform chemicals have become an alternative feedstock for chemical industry. They may replace conventional fossil-based resources in two senses. On one hand biomass based compounds can be used for the synthesis of already existing products in the chemical industry as they are available mainly from petroleum. On the other hand, these biomass or biomass-derived compounds can be used to produce new and original compounds, which is facilitated by characteristic structure elements such as the presence of numerous hydroxyl functions in carbohydrates. Photochemical or photocatalytic reactions make compounds or compound families available that cannot or difficultly synthesized using conventional methods of organic synthesis. A high degree of molecular complexity and diversity is thus generated in a facile way. This is explained by the complementarity of ground state and excited state reactivity. In this same context, it should be pointed out that in the past, photochemical or photocatalytic reactions of biomass-derived products have rarely been investigated in a systematic way.
Photochemical and catalytic reactions on one hand and the utilization of biomass as a renewable feedstock on the other are part of sustainable chemistry. The combination of both opens perspectives for sustainable chemistry.
References
- Anastas, P.T.; Kirchhoff, M.M. Origins, Current Status, and Future Challenges of Green Chemistry. ACC Chem. Res. 2002, 35, 686–694.
- Wertz, J.-L.; Bédué, O. Lignocellulosic Biorefineries; EPFL Press: Lausanne, Switzerland, 2013.
- Barrault, J.; Kervennal, J.; Isnard, P. La chimie durable: Pour l’environement, l’économie notre socciété (Numéro thématique). Actual. Chim. 2018, 15–116.
- Marion, P.; Bernela, B.; Piccirilli, A.; Estrine, B.; Patoullard, N.; Guilbot, J.; Jérôme, F. Sustainable chemistry: How to produce better and more from less? Green Chem. 2017, 19, 4973–4989.
- Lichtenthaler, F.W.; Peters, S. Carbohydrates as green raw materials for the chemical industry. Comptes Rendus Chim. 2004, 7, 65–90.
- Lichtenthaler, F.W. Carbohydrate-based Product Lines. In Biorefineries–Industrial Processes and Products; Kamm, B., Gruber, P.R., Kamm, M., Eds.; Wiley-VCH: Weinheim, Germany, 2006; Volume 1, pp. 2–59.
- Heitner, C.; Dimmel, D.R.; Schmidt, J.A. (Eds.) Lignin and Lignans; CRC Press: Boca Raton, FL, USA, 2010.
- Farmer, T.J.; Mascal, M. Platfom molecules. In Introduction to Chemicals from Biomass, 2nd ed.; Clark, J., Deswarte, F., Eds.; Wiley: Chichester, UK, 2015; pp. 89–152.
- Cramail, H.; Bizet, B.O.; Durand, P.-L.; Hibert, G.; Grau, E. Bio-Sourced Polymers: Recent Advances In Avanced Green Chemistry, Part 2; Horváth, I.T., Malacria, M., Eds.; World Scientific: Singapore, 2020; pp. 167–328.
- Esposito, D.; Antonietti, M. Redefining biorefinery: The search for unconventional building blocks for materials. Chem. Soc. Rev. 2015, 44, 5821–5835.
- Monet, E. La forêt: Un gisement privilege et durable de molecules biosourcées. Actual. Chim. 2023, 14–23.
- de Rezende Locatel, W.; Guilhaume, N.; Schuurman, Y.; Laurenti, D. La pyrolyse du bois et la conversion catalytique de ses vapeurs. Actual. Chim. 2023, 71–77.
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558.
- Tuck, C.O.; Pérez, E.; Horvath, I.; Sheldon, R.A.; Poliakoff, M. Valorization of Biomass: Deriving More Value from Waste. Science 2012, 337, 695–699.
- Ravelli, D.; Samorì, C. (Eds.) Biomass Valorisation; Wiley-VCH: Weinheim, Germany, 2021.
- Corma, A.; Iborra, S.; Velty, A. Chemical Routes for the Transformation of Biomass into Chemicals. Chem. Rev. 2007, 107, 2411–2502.
- Chatterjee, C.; Pong, F.; Sen, A. Chemical conversion pathways for carbohydrates. Green Chem. 2015, 17, 40–71.
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Bimetallic catalysts for upgrading of biomass to fuels and chemicals. Chem. Soc. Rev. 2012, 41, 8075–8098.
- Groß, J.; Kühlborn, J.; Opatz, T. Applications of xylochemistry from laboratory to industrial scale. Green Chem. 2020, 22, 4411–4425.
- Kühlborn, J.; Groß, J.; Opatz, T. Making natural products from renewable feedstocks: Back to the roots? Nat. Prod. Rep. 2020, 37, 380–424.
- Sheldon, R.A.; Arends, I.; Hanefeld, U. (Eds.) Green Chemistry and Catalysis; Wiley-VCH: Weinheim, Germany, 2007.
- Centi, G.; van Santen, R.A. (Eds.) Catalysis for Renewables; Wiley-VCH: Weinheim, Germany, 2007.
- Imhof, P.; van der Waal, J.C. (Eds.) Catalytic Process Development for Renewable Materials; Wiley-VCH: Weinheim, Germany, 2013.
- Protti, S.; Manzini, S.; Fagnoni, M.; Albini, A. RSC Green Chemistry Book Series, Eco-Friendly Synthesis of Fine Chemicals; Ballini, R., Ed.; Royal Society of Chemistry: London, UK, 2009; pp. 80–111.
- Ciamician, G. The Photochemistry of the Future. Science 1912, 36, 385–394.
- Ciamician, G. Sur les actions de la lumière. Bull. Soc. Chim. Fr. 1908, 3, i.
- Hoffmann, N. Photochemical reactions of aromatic compounds and the concept of the photon as a traceless reagent. Photochem. Photobiol. Sci. 2012, 11, 1613–1641.
- Oelgemöller, M.; Jung, C.; Mattay, J. Green photochemistry: Production of fine chemicals with sunlight. Pure Appl. Chem. 2007, 79, 1939–1947.
- Turro, N.J.; Schuster, G. Photochemical Reactions as a Tool in Organic Syntheses. Science 1975, 187, 303–312.
- Hoffmann, N. Photochemical Key Steps in organic Synthesis. Chem. Rev. 2008, 108, 1052–1103.
- Bach, T.; Hehn, J.P. Photochemical Reactions as Key Steps in Natural Product Synthesis. Angew. Chem. Int. Ed. 2011, 50, 1000–1052.
- Kärkäs, M.D.; Porco, J.A.; Stephenson, C.R.J. Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev. 2016, 116, 9683–9747.
- Bonfield, H.E.; Knauber, T.; Lévesque, F.; Moschetta, E.G.; Susanne, F.; Edwards, L.J. Photons as a 21st century reagent. Nat. Commun. 2020, 11, 804.
- Lefebvre, C.; Fortier, L.; Hoffmann, N. Photochemical Rearrangements in Heterocyclic Chemistry. Eur. J. Org. Chem. 2020, 2020, 1393–1404.
- Latrache, M.; Hoffmann, N. Photochemical radical cyclization reactions with imines, hydrazones, oximes and related compounds. Chem. Soc. Rev. 2021, 50, 7418–7435.
- Hoffmann, N. Enantioselective synthesis of heterocyclic compounds using photochemical reactions. Photochem. Photobiol. Sci. 2021, 20, 1657–1674.
- Michelin, C.; Hoffmann, N. Photocatalysis applied to organic synthesis—A green chemistry approach. Curr. Opin. Green Sustain. Chem. 2018, 10, 40–45.
- Michelin, C.; Hoffmann, N. Photosensitization and Photocatalysis—Perspectives in Organic Synthesis. ACS Catal. 2018, 8, 12046–12055.
- Tyburski, R.; Liu, T.; Glover, S.D.; Hammarström, L. Proton-Coupled Electron Transfer Guidelines, Fair and Square. J. Am. Chem. Soc. 2021, 143, 560–576.
- Miller, D.C.; Tarantino, K.T.; Knowles, R.R. Proto-Coupled Electron Transfer in Organic Synthesis: Fundamentals, Applications, and Opportunities. Top. Curr. Chem. 2016, 374, 30.
- Hoffmann, N. Proton-Coupled Electron Transfer in Photoredox Catalytic Reactions. Eur. J. Org. Chem. 2017, 2017, 1982–1992.
- Stephenson, C.R.J.; Yoon, T.P.; MacMillan, D.W.C. (Eds.) Visible Light Photocatalysis in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2018.
- König, B. (Ed.) Chemical Photocatalysis, 2nd ed.; De Gruyter: Berlin, Germany, 2020.
- Marzo, L.; Pagire, S.K.; Reiser, O.; König, B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072.
- Barata-Vallejo, S.; Yerien, D.E.; Postigo, A. Bioinspired Photocatalyzed Organic Synthetic Transformations. The Use of Natural Pigments and Vitamins in Photocatalysis. ChemCatChem 2022, e202200623.
- Mattay, J. Charge Transfer and Radical Ions in Photochemistry. Angew. Chem. Int. Ed. 1987, 26, 825–845.
- Zeitler, K.; Neumann, M. Synergistic visible light photoredox catalysis. In Chemical Photocatalysis, 2nd ed.; König, B., Ed.; De Gruyter: Berlin, Germany, 2020; pp. 245–283.
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074.
- Hopkinson, M.N.; Sahoo, B.; Li, J.-L.; Glorius, F. Dual Catalysis Sees the Light: Combining Photoredox with Organo-, Acid, and Transition-Metal Catalysis. Chem. Eur. J. 2014, 20, 3874–3886.
- Ooi, T. Molecular Technology: Energy Innovation; Yamamoto, H., Kato, T., Eds.; Wiley-VCH: Weinheim, Germany, 2018; pp. 131–163.
- Mastandrea, M.M.; Pericàs, M. Photoredox Dual Catalysis: A Fertile Playground for the Discovery of New reactivities. Eur. J. Inorg. Chem. 2021, 2021, 3421–3431.
- Lévêque, C.; Levernier, E.; Corcé, V.; Fensterbank, L.; Malacria, M.; Ollivier, C. Photoredox Catalysis, an Opportunity for Sustainable Radical Chemistry. In Avanced Green Chemistry, Part 2; Horváth, I.T., Malacria, M., Eds.; World Scientific: Singapore, 2020; pp. 49–121.
- Yang, N.; Tian, Y.; Zhang, M.; Peng, X.; Li, F.; Li, J.; Li, Y.; Fan, B.; Wang, F.; Song, H. Photocatalyst-enzyme hybrid systems for light-driven biotransformations. Biotechnol. Adv. 2022, 54, 107808.
- Lee, S.H.; Choi, D.S.; Kuk, S.K.; Park, C.B. Photobiocatalysis: Activating Redox Enzymes by Direct or Indirect Transfer of Photoinduced Electrons. Angew. Chem. Int. Ed. 2018, 57, 7958–7985.
- Rudorff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 2018, 1, 12–22.
- Schmermund, L.; Jurkas, V.; Özgen, F.F.; Barone, G.D.; Büchsenschütz, H.C.; Winkler, C.K.; Schmidt, S.; Kourist, R.; Kroutil, W. Photo-Biocatalysis: Biotransformations in the Resence of Light. ACS Catal. 2019, 9, 4115–4144.
- Zhang, W.; Hollmann, F. Noncoventional regeneration of redox enzymes—A practical approach for organic synthesis. Chem. Commun. 2018, 54, 7281–7289.
- Nicewicz, D.A.; MacMillan, D.W.C. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80.
- Saha, D. Catalytic Enantioselective Radical Transformations Enabled by Visible Light. Chem. Asian J. 2020, 15, 2129–2152.
- Brimioulle, R.; Lenhart, D.; Maturi, M.M.; Bach, T. Enantioselective Catalysis of Photochemical Reactions. Angew. Chem. Int. Ed. 2015, 54, 3872–3890.
- Jiang, C.; Chen, W.; Zheng, W.-H.; Lu, H. Advances in asymmetric visible-light photocatalysis, 2015–2019. Org. Biomol. Chem. 2019, 17, 8673–8689.
- Fidaly, K.; Ceballos, C.; Falguières, A.; Sylla-Iyarreta Veitia, M.; Guy, A.; Ferroud, C. Visible light photoredox organocatalysis: A fully transition metal-free direct asymmetric α-alkylation of aldehydes. Green Chem. 2012, 14, 1293–1297.
- Cheung, K.P.S.; Sarkar, S.; Gevorgyan, V. Visible Light-Induced Transition Metal Catalysis. Chem. Rev. 2022, 122, 1543–1625.
- Lipp, A.; Badir, S.O.; Molander, G.A. Stereoinduction in Metallaphotoredox Catalysis. Angew. Chem. Int. Ed. 2021, 60, 1714–1726.
- Hoffmann, N. Combining Photoredox and Metal Catalysis. ChemCatChem 2015, 7, 393–394.
- Fabry, D.C.; Rueping, M. Merging Visible Light Photoredox Catalysis with Metal Catalyzed C-H Activations: On the Role of Oxygen and Superoxide Ions as Oxidants. ACC Chem. Res. 2016, 49, 1969–1979.
- Sadek, O.; Abdellaoui, M.; Millanvois, A.; Ollivier, C.; Fensterbank, L. Organometallic catalysis under visible light activation: Benefits and preliminary rationals. Photochem. Photobiol. Sci. 2022, 21, 585–606.
- Huo, H.; Shen, X.; Wang, C.; Zhang, L.; Röse, P.; Chen, L.A.; Harms, K.; Marsch, M.; Hilt, G.; Meggers, E. Asymmetric photoredox transition-metal catalysis ativated by visible light. Nature 2014, 515, 100–103.
- Wu, X.; Luo, N.; Xie, S.; Zhang, H.; Zhang, Q.; Wang, F.; Wang, Y. Photocatalytic transformations of lignocellulosic biomass into chemicals. Chem. Soc. Rev. 2020, 49, 6198–6223.
- Colmenares, J.C.; Luque, R. Heterogeneous photocatalytic nanomaterials: Prospects and challenges in selective transformations of biomass-derived compounds. Chem. Soc. Rev. 2014, 43, 765–778.
- Granone, L.I.; Sieland, F.; Zheng, N.; Dillert, R.; Bahnemann, D.W. Photocatalytic conversion of biomass into valuable products: A meaningful approach? Green Chem. 2018, 20, 1169–1192.
- Rao, V.N.; Malu, T.J.; Cheralathan, K.K.; Sakar, M.; Pitchaimuthu, S.; Rodríguez-González, V.; Kumari, M.M.; Shankar, M.V. Light-driven transformation of biomass into chemicals using photocatalysts—Vistas and challenges. J. Environ. Manag. 2021, 284, 111983.
- Fan, H.; Li, G.; Yang, F.; Yang, L.; Zhang, S. Photodegradation of cellulose under UV light catalysed by TiO2. J. Chem. Technol. Biotechnol. 2011, 86, 1107–1112.
- Wang, L.; Zhang, Z.; Zhang, L.; Xue, S.; Doherty, W.O.S.; O’Hara, I.M.; Ke, X. Sustainable conversion of cellulosic biomass to chemicals under visible-light irradiation. RSC Adv. 2015, 5, 85242–85247.
- Laugel, C.; Estrine, B.; Le Bras, J.; Hoffmann, N.; Marinkovic, S.; Muzart, J. Visible Light-Accelerated Depolymerisation of Starch under Fenton Conditions and Preparation of Calcium Sequestering Compounds. Catal. Lett. 2014, 144, 1674–1680.
- Li, S.-H.; Liu, S.; Colmenares, J.C.; Xu, Y.-J. A sustainable approach for lignin valorization by heterogeneous photocatalysis. Green Chem. 2016, 18, 594–607.
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599.
- Rahimi, A.; Ulbrich, A.; Coon, J.J.; Stahl, S.S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 2014, 515, 249–252.
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. Int. Ed. 2016, 55, 8164–8215.
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905.
- Nayak, J.; Basu, A.; Dey, P.; Kumar, R.; Upadhaya, A.; Ghosh, S.; Bishayee, B.; Mishra, S.R.; Tripathy, S.K.; Banerjee, S.; et al. Transformation of agro-biomass into vanillin through novel membrane integrated value-addition process: A state-of-the-art review. Biomass Conv. Bioref. 2022, 1–24.
- Kärkäs, M.D.; Matsuura, B.S.; Monos, T.M.; Magallanes, G.; Stephenson, C.R.J. Transition-metal catalyzed valorization of lignin: The key to a sustainable carbon-neutral future. Org. Biomol. Chem. 2016, 14, 1853–1914.
- Magallanes, G.; Kärkäs, M.D.; Bosque, I.; Lee, S.; Maldonado, S.; Sephenson, C.R.J. Selective C–O Bond Cleavage of Lignin Systems and Polymers Enabled by Sequential Palladium-Catalyzed Aerobic Oxidation and Visible-Light Photoredox Catalysis. ACS Catal. 2019, 9, 2252–2260.
- Muzart, J. Palladium-catalysed oxidation of primary and secondary alcohols. Tetrahedron 2003, 59, 5789–5816.
- Hoffmann, N. Electron and hydrogen transfer in organic photochemical reactions. J. Phys. Org. Chem. 2015, 28, 121–136.
- Zhang, J. Conversion of Lignin Models by Photoredox Catalysis. ChemSusChem 2018, 11, 3071–3080.
- Das, A.; König, B. Transition metal- and photoredox-catalyzed valorisation of lignin subunits. Green Chem. 2018, 20, 4844–4852.
- Shatskiy, A.; Kärkäs, M.D. Biomass Processing via Photochemical Means. In Biomass Valorisation; Ravelli, D., Samorì, C., Eds.; Wiley-VCH: Weinheim, Germany, 2021; pp. 265–288.
- Martín-Perales, A.I.; Rodríguez-Padron, D.; García Coleto, A.; Len, C.; de Miguel, G.; Muñoz-Batista, M.J.; Rafael Luque, R. Photocatalytic Production of Vanilline over CeOx and ZrO2 Modified Biomass-Templated Titania. Ind. Eng. Chem. Res. 2020, 59, 17085–17093.
- Camera-Roda, G.; Augugliaro, V.; Cardillo, A.; Loddo, V.; Palmisano, G.; Palmisano, L. A pervaporation reactor for the green Synthesis of Vanillin. Chem. Eng. J. 2013, 224, 136–143.
- Desvals, A.; Baudron, S.A.; Bulach, V.; Hoffmann, N. Photocycloadditions of Arenes Derived from Lignin. J. Org. Chem. 2021, 86, 13310–13321.
- Desvals, A.; Hoffmann, N. Photocycloadditions of benzene derivatives and their systematic application to organic synthesis. Aus. J. Chem. 2023, 76, 117–129.
- Remy, R.; Bochet, C.G. Arene-Alkene Cycloaddition. Chem. Rev. 2016, 116, 9816–9849.
- Chapleur, Y.; Chrétien, F. Sugars as Chiral Starting Materials in Enantiospecific Synthesis. In The Organic Chemistry of Sugars; Levy, D.E., Fügedi, P., Eds.; CRC Taylor & Francis: Boca Raton, FL, USA, 2006; pp. 489–573.
- Vogel, P. Total Asymmetric Synthesis of Monosaccharides and Analogs. In The Organic Chemistry of Sugars; Levy, D.E., Fügedi, P., Eds.; CRC Taylor & Francis: Boca Raton, FL, USA, 2006; pp. 629–725.
- Lichtenthaler, F.W. Emil Fischer’s Proof of the Configuration of Sugars: A Centennial Tribute. Angew. Chem. Int. Ed. 1992, 31, 1541–1556.
- Stick, R.V. Carbohydrates—The Sweet Molecules of Life; Academic Press: San Diego, CA, USA, 2001.
- Toshima, K. Synthesis of Carbohydrate Containing Complex Natural Compounds. In The Organic Chemistry of Sugars; Levy, D.E., Fügedi, P., Eds.; CRC Taylor & Francis: Boca Raton, FL, USA, 2006; pp. 575–627.
- Kuszmann, J. Ingtoduction to Carbohydrates. In The Organic Chemistry of Sugars; Levy, D.E., Fügedi, P., Eds.; CRC Taylor & Francis: Boca Raton, FL, USA, 2006; pp. 25–52.
- Shatskiy, A.; Stepanova, E.V.; Kärkäs, M.D. Exploiting photoredox catalysis for carbohydrate modification through C-H and C-C activation. Nat. Rev. Chem. 2022, 6, 782–805.
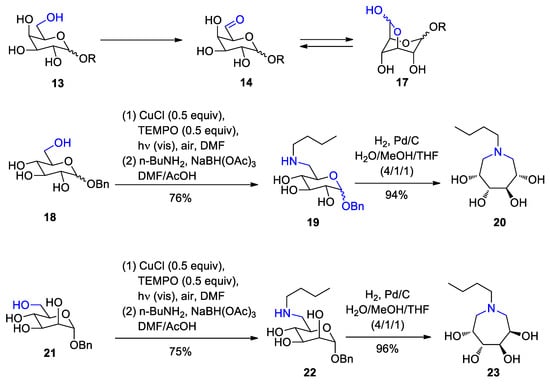
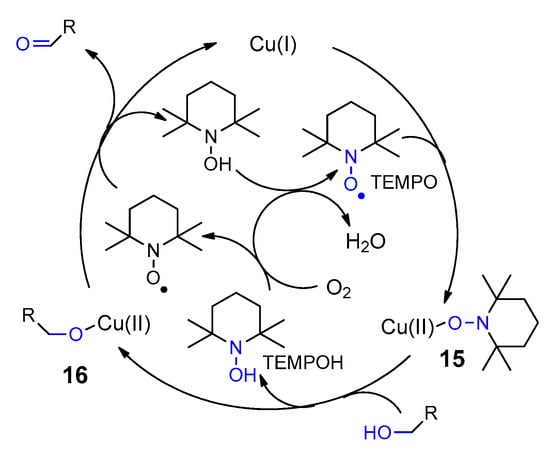
- Gassama, A.; Hoffmann, N. Selective Light Supported Oxidation of Hexoses Using Air as Oxidant—Synthesis of Tetrahydroxyazepanes. Adv. Synth. Catal. 2008, 350, 35–39.
- Ito, N.; Phillips, S.E.V.; Stevens, C.; Ogel, Z.B.; McPhersonn, M.J.; Keen, J.N.; Yadav, K.D.S.; Knowles, P.F. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature 1991, 350, 87–90.
- Thomas, F. Ten Years of a Biomimetic Approach to the Copper(II) Radical Site of Galactose Oxidase. Eur. J. Inorg. Chem. 2007, 2007, 2379–2404.
- Pierre, J.-L. One electron at a time oxidations and enzymatic paradigms: From metallic to non-metallic redox centers. Chem. Soc. Rev. 2000, 29, 251–257.
- Berkessel, A.; Dousset, M.; Bulat, S.; Glaubitz, K. Combinatorial approaches to functional models for galactose oxidase. Biol. Chem. 2005, 386, 1035–1041.
- Chaudhuri, P.; Hess, M.; Hildenbrand, K.; Bill, E.; Weyhermüller, T.; Wieghardt, K. Aerobic Oxidation of Primary Alcohols (Including Methanol) by Copper(II)− and Zinc(II)−Phenoxyl Radical Catalysts. J. Am. Chem. Soc. 1999, 121, 9599–9610.
- Semmelhack, M.F.; Schmid, C.R.; Cortés, D.A.; Chou, C.S. Oxidation of alcohols to aldehydes with oxygen and cupric ion, mediated by nitrosonium ion. J. Am. Chem. Soc. 1984, 106, 3374–3376.
- Ryland, B.L.; Stahl, S.S. Practical Aerobic Oxidations of Alcohols and Amines with Homogeneous Copper/TEMPO and Related Catalyst Systems. Angew. Chem. Int. Ed. 2014, 53, 8824–8838.
- Dijksman, A.; Arends, I.W.C.E.; Sheldon, R.A. Cu(ii)-nitroxyl radicals as catalytic galactose oxidase mimics. Org. Biomol. Chem. 2003, 1, 3232–3237.
- Abderrazak, Y.; Bhattacharyya, A.; Reiser, O. Visible-Light-Induced Homolysis of Earth-Abundant Metal-Substrate Complexes: A Complementary Activation Strategy in Photoredox Catalysis. Angew. Chem. Int. Ed. 2021, 60, 21100–21115.
- Compain, P.; Martin, O.R. (Eds.) Iminosugars; Wiley & Sons: Chichester, UK, 2007.
- Li, H.; Marcelo, F.; Bello, C.; Vogel, P.; Butters, T.D.; Rauter, A.P.; Zhang, Y.; Sollogoub, M.; Blériot, Y. Design and synthesis of acetamido tri- and tetra-hydroxyazepanes: Potent and selective β-N-acetylhexosaminidase inhibitors. Bioorg. Med. Chem. 2009, 17, 5598–5604.
- Joseph, C.C.; Regeling, H.; Zwanenburg, B.; Chittenden, G.J.F. Syntheses of (3R,4R,5R,6R)-tetrahydroxyazepane (1,6-dideoxy-1,6-imino-d-mannitol) and (3S,4R,5R,6R)-tetrahydroxyazepane (1,6-dideoxy-1,6-imino-d-glucitol). Tetrahedron 2002, 58, 6907–6911.
- Oelgemöller, M. Solar Photochemical Synthesis: From the Beginnings of Organic Photochemistry to the Solar Manufacturing of Commodity Chemicals. Chem. Rev. 2016, 116, 9664–9682.
- Sangwan, R.; Mandal, P.K. Recent advances in photoinduced glycosylation: Oligosaccharides, glycoconjugates and their synthetic applications. RSC Adv. 2017, 7, 26256–26321.
- Ling, J.; Bennett, C.S. Recent Developments in Stereoselective Chemical Glycosylation. Asian J. Org. Chem. 2019, 8, 802–813.
- Fügedi, P. Glycosylation Methods. In The Organic Chemistry of Sugars; Levy, D.E., Fügedi, P., Eds.; CRC Taylor & Francis: Boca Raton, FL, USA, 2006; pp. 89–179.
- Collins, P.; Ferrier, R. Monosaccharides; Wiley: Chichester, UK, 1995.
- Hashimoto, S.; Kurimoto, I.; Fujii, Y.; Noyori, R. Novel nucleophilic substitution reaction by radical cation intermediates. Photosensitized transacetalization via SON1 mechanism. J. Am. Chem. Soc. 1985, 107, 1427–1429.
- Wever, W.J.; Cinelli, M.A.; Bowers, A.A. Visible Light Mediated Activation and O-Glycosylation of Thioglycosides. Org. Lett. 2013, 15, 30–33.
- Tolbert, L.M.; Solntsev, K.M. Excited-State Proton Transfer: From Constrained Systems to “Super” Photoacids to Superfast Proton Transfer. ACC Chem. Res. 2002, 35, 19–27.
- Zivic, N.; Kuroishi, P.K.; Dumur, F.; Gigmes, D.; Dove, A.P.; Sardon, H. Recent Advances and Challenges in the Design of Organic Photoacid and Photobase Generators for Polymerizations. Angew. Chem. Int. Ed. 2019, 58, 10410–10422.
- Saway, J.; Salem, Z.M.; Badillo, J.J. Recent Advances in Photoacid Catalysis for Organic Synthesis. Synthesis 2021, 53, 489–497.
- Iwata, R.; Uda, K.; Takahashi, D.; Toshima, K. Photo-induced glycosylation using reusable organophotoacids. Chem. Commun. 2014, 50, 10695–10698.
- Kimura, T.; Eto, T.; Takahashi, D.; Toshima, K. Stereocontrolled Photoinduced Glycosylation Using an Aryl Thiourea as an Organo photoacid. Org. Lett. 2016, 18, 3190–3193.
- Zhao, G.; Li, J.; Wang, T. Visible-light-induced photoacid catalysis: Application in glycosylation with O-glycosyl trichloroacetimidates. Chem. Commun. 2021, 57, 12659–12662.
- Levy, D.E. Strategies towards C-Glycosides. In The Organic Chemistry of Sugars; Levy, D.E., Fügedi, P., Eds.; CRC Taylor & Francis: Boca Raton, FL, USA, 2006; pp. 269–348.
- Zhang, M.; Kong, L.; Gong, R.; Iorio, M.; Donatio, S.; Deng, Z.; Sosio, M.; Chen, W. Biosynthesis of C-nucleoside antibiotics in actinobacteria: Recent advances and future developments. Microbiol. Cell Factories 2022, 21, 2.
- Zhu, M.; Messoudi, S. Diastereoselective Decarboxylative Alkynylation of Anomeric Carboxylic Acids Using Cu/Photoredox Dual Catalysis. ACS Catal. 2021, 11, 6334–6342.
- Schwarz, J.; König, B. Decarboxylative reactions with and without light—A comparison. Green Chem. 2018, 20, 323–361.
- Martinez, R.D.; Buitrago, A.A.; Howell, N.W.; Hearn, C.H.; Joens, J.A. The near UV absorption spectra of several aliphatic aldehydes and ketones at 300 K. Atmos. Environ. 1992, 26A, 785–792.
- Jahjah, R.; Gassama, A.; Bulach, V.; Suzuki, C.; Abe, M.; Hoffmann, N.; Martinez, A.; Nuzillard, J.-M. Stereoselective Triplet-Sensitised Radical Reactions of Furanone Derivatives. Chem. Eur. J. 2010, 16, 3341–3354.
- Oelgemöller, M.; Hoffmann, N.; Shvydkiv, O. From ‘Lab & Light on a Chip’ to Parallel Microflow Photochemistry. Aust. J. Chem. 2014, 67, 337–342.
- Hoffmann, N. Photochemical Electron and Hydrogen Transfer in Organic Synthesis: The Control of Selectivity. Synthesis 2016, 48, 1782–1802.
- Wessig, P.; Mühling, O. Abstraction of (γ ± n)-Hydrogen by Excited Carbonyls. In Synthetic Organic Photochemistry; Giesbeck, A.G., Mattay, J., Eds.; Marcel Dekker: New York, NY, USA, 2005; pp. 41–87.
- Martín, A.; Suárez, E. Carbohydrate Spiro-heterocycles via Radical Chemistry. Top. Heterocycl. Chem. 2019, 57, 51–104.
- Thiering, S.; Sund, C.; Thiem, J.; Giesler, A.; Kopf, J. Syntheses of imido-substituted glycosans and their photocyclisation towards highly functionalised heterotricycles. Carbohydr. Res. 2001, 336, 271–282.
- Capaldo, L.; Ravelli, D. Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2017, 2056–2071.
- Ravelli, D.; Protti, S.; Fagnoni, M. Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913.
- Fréneau, M.; de Sainte-Claire, P.; Abe, M.; Hoffmann, N. The structure of electronically excited α,β-unsaturated lactones. J. Phys. Org. Chem. 2016, 29, 718–724.
- Lejeune, G.; Font, J.; Parella, T.; Alibés, R.; Figueredo, M. Intramolecular Photoreactions of (5S)-5-Oxymethyl-2(5H)-furanones as a Tool for the Stereoselective Generation of Diverse Polycyclic Scaffolds. J. Org. Chem. 2015, 80, 9437–9445.
- Tabanelli, T.; Cavani, F. Industrial Perspectives of Biomass Processing. In Biomass Valorisation; Ravelli, D., Samorì, C., Eds.; Wiley-VCH: Weinheim, Germany, 2021; pp. 369–410. [Google Scholar]
- Welter, R.A.; Santana, H.; de la Torre, L.G.; Robertson, M.; Taranto, O.P.; Oelgemöller, M. Methyl Oleate Synthesis by TiO2 Photocatalytic Esterification of Oleic Acid: Optimisation by Response Surface Quadratic Methodology, Reaction Kinetics and Thermodynamics. ChemPhotoChem 2022, 6, e202200007. [Google Scholar] [CrossRef]
- Huang, J.; Jian, Y.; Zhu, P.; Abdelaziz, O.; Li, H. Research Progress on the Photo-Driven Catalytic Production of Biodiesel. Front. Chem. 2022, 10, 904251. [Google Scholar] [CrossRef]
- Belousov, A.S.; Suleimanov, E.V. Application of metal–organic frameworks as an alternative to metal oxide-based photocatalysts for the production of industrially important organic chemicals. Green Chem. 2021, 23, 6172–6204. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, G.; Sun, P. Transition metal-free decarboxylative alkylation reactions. Org. Biomol. Chem. 2016, 14, 10763–10777. [Google Scholar] [CrossRef]
- Schwarz, J. Photocatalytic decarboxylations. Phys. Sci. Rev. 2018, 3, 20170186. [Google Scholar] [CrossRef]
- Tabandeh, M.; Cheng, C.K.; Centi, G.; Show, P.L.; Chen, W.-H.; Ling, T.C.; Ong, H.C.; Ng, E.-P.; Juan, J.C.; Lam, S.S. Recent advancement in deoxygenation of fatty acids via homogeneous catalysis for biofuel production. Mol. Catal. 2022, 523, 111207. [Google Scholar] [CrossRef]
- Patra, T.; Maiti, D. Decarboxylation as the Key Step in C−C Bond-Forming Reactions. Chem. Eur. J. 2017, 23, 7382–7401. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Kramer, W.; Oelgemöller, M. Photoinduced decarboxylation reactions. Radical chemistry in water. Green Chem. 1999, 1, 205–208.
- Maeda, K.; Saito, H.; Osaka, K.; Nishikawa, K.; Sugie, M.; Morita, T.; Takahashi, I.; Yoshimi, Y. Direct modification of tripeptides using photoinduced decarboxylative radical reactions. Tetrahedron 2015, 71, 1117–1123.
- Kicatt, D.M.; Nicolle, S.; Lee, A.-L. Direct decarboxylative Giese reactions. Chem. Soc. Rev. 2022, 51, 1415–1453.
- Rodríguez, N.; Goossen, L.J. Decarboxylative coupling reactions: A modern strategy for C–C-bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048.
- McMurray, L.; McGuire, T.M.; Howells, R.L. Recent Advances in Photocatalytic Decarboxylative Coupling Reactions in Medicinal Chemistry. Synthesis 2020, 52, 1719–1737.
- Yoshimi, Y.; Itou, T.; Hatanaka, M. Decarboxylative reduction of free aliphatic carboxylic acids by photogenerated cation radical. Chem. Comm. 2007, 5244–5246.
- Griffin, J.D.; Zeller, M.A.; Nicewicz, D.A. Hydrodecarboxylation of Carboxylic and Malonic Acid Derivatives via Organic Photoredox Catalysis: Substrate Scope and Mechanistic Insight. J. Am. Chem. Soc. 2015, 137, 11340–11348.
- Huang, Z.; Zhao, Z.; Zhang, C.; Liu, J.; Luo, N.; Zhang, J.; Wang, F. Enhanced photocatalytic alkane production from fatty acid decarboxylation via inhibition of radical oligomerization. Nat. Catal. 2020, 3, 170–178.
- Sorigué, D.; Légeret, B.; Cuiné, S.; Blangy, S.; Moulin, S.; Billon, E.; Richaud, P.; Brugière, S.; Couté, Y.; Nurizzo, D.; et al. An algal photoenzyme converts fatty acids to hydrocarbons. Science 2017, 357, 903–907.
- Sorigué, D.; Hadjidemetriou, K.; Blangy, S.; Gotthard, G.; Bonvalet, A.; Coquelle, N.; Samire, P.; Aleksandrov, A.; Antonucci, L.; Benachir, A.; et al. Mechanism and dynamics of fatty acid photodecarboxylase. Science 2021, 372, eabd5687.
- Harrison, W.; Huang, X.; Zhao, H. Photobiocatalysis for Abiological Transformations. Acc. Chem. Res. 2022, 55, 1087–1096.
- Amer, M.; Wojcik, E.Z.; Sun, C.; Hoeven, R.; Hughes, J.M.X.; Faulkner, M.; Yunus, I.S.; Tait, S.; Johannissen, L.O.; Hardman, S.J.O.; et al. Low carbon strategies for sustainable bio-alkane gas production and renewable energy. Energy Environ. Sci. 2020, 13, 1818–1831.
- Zhang, W.; Ma, M.; Huijbers, M.M.E.; Filonenko, G.A.; Pidko, E.A.; van Schie, M.; de Boer, S.; Burek, B.O.; Bloh, J.Z.; van Berkel, W.J.H.; et al. Hydrocarbon Synthesis via Photoenzymatic Decarboxylation of Carboxylic Acids. J. Am. Chem. Soc. 2019, 141, 3116–3120.
- Duong, H.T.; Wu, Y.; Sutor, A.; Burek, B.O.; Hollmann, F.; Bloh, J.Z. Intensification of Photobiocatalytic Decarboxylation of Fatty Acids for the Production of Biodiesel. ChemSusChem 2021, 14, 1053–1056.
- Griesbeck, A.G.; Kramer, W.; Oelgemöller, M. Synthetic Applications of Photoinduced Electron Transfer Decarboxylation Reactions. Synlett 1999, 1169–1178.
- Xuan, J.; Zhang, Z.G.; Xiao, W.J. Visible-Light-Induced Decarboxylative Functionalization of Carboxylic Acids and Their Derivatives. Angew. Chem. Int. Ed. 2015, 54, 15632–15641.
- Zheng, Y.; Shao, X.; Ramadoss, V.; Tian, L.; Wang, Y. Recent Developments in Photochemical and Electrochemical Decarboxylative C(sp3)–N Bond Formation. Synthesis 2020, 52, 1357–1368.
- Mumtaz, S.; Robertson, M.J.; Oelgemöller, M. Recent advances in photodecarboxylations involving phthalimides. Aust. J. Chem. 2018, 71, 634–648.
- Jin, Y.; Fu, H. Visible-Light Photoredox Decarboxylative Couplings. Asian J. Org. Chem. 2017, 6, 368–385.
- De bruyn, M.; Fan, J.; Budarin, V.L.; Macquarrie, D.J.; Gomez, L.D.; Simister, R.; Farmer, T.J.; Raverty, W.D.; McQueen-Mason, S.J.; Clark, J.H. A new perspective in bio-refining: Levoglucosenone and cleaner lignin from waste biorefinery hydrolysis lignin by selective conversion of residual saccharides. Energy Environ. Sci. 2016, 9, 2571–2574. [Google Scholar] [CrossRef][Green Version]
- Halpern, Y.; Ritter, R.; Broido, A. Levoglucosenone (1,6-anhydro-3,4-dideoxy-.DELTA.3-.beta.-D-pyranosen-2-one). Major product of the acid-catalyzed pyrolysis of cellulose and related carbohydrates. J. Org. Chem. 1973, 38, 204–209. [Google Scholar] [CrossRef]
- He, J.; Liu, M.; Huang, K.; Walker, T.W.; Maravelias, C.T.; Dumesic, J.A.; Huber, G.W. Production of levoglucosenone and 5-hydroxymethylfurfural from cellulose in polar aprotic solvent–water mixtures. Green Chem. 2017, 19, 3642–3653. [Google Scholar] [CrossRef]
- Sherwood, J.; De bruyn, M.; Constantinou, A.; Moity, L.; McElroy, C.R.; Farmer, T.J.; Duncan, T.; Raverty, W.; Hunt, A.J.; Clark, J.H. Dihydrolevoglucosenone (Cyrene) as a bio-based alternative for dipolar aprotic solvents. Chem. Commun. 2014, 50, 9650–9652. [Google Scholar] [CrossRef]
- Comba, M.B.; Tsai, Y.; Sarotti, A.M.; Mangione, M.I.; Suárez, A.G.; Spanevello, R.A. Levoglucosenone and Its New Applications: Valorization of Cellulose Residues. Eur. J. Org. Chem. 2018, 2018, 590–604. [Google Scholar] [CrossRef]
- Awad, L.; Demange, R.; Zhu, Y.-H.; Vogel, P. The use of levoglucosenone and isolevoglucosenone as templates for the construction of C-linked disaccharides. Carbohydr. Res. 2006, 341, 1235–1252. [Google Scholar] [CrossRef] [PubMed]
- Sarotti, A.M.; Zanardi, M.M.; Spanevello, R.A.; Suárez, A.G. Recent Applications of Levoglucosenone as Chiral Synthon. Curr. Org. Synth. 2012, 9, 439–459. [Google Scholar] [CrossRef]
- Tsai, Y.; Borini Etichetti, C.M.; Di Benedetto, C.; Girardini, J.E.; Terra Martins, F.; Spanevello, R.A.; Suárez, A.; Sarotti, A.M. Synthesis of Triazole Derivatives of Levoglucosenone as Promising Anticancer Agents: Effective Exploration of the Chemical Space through retro-aza-Michael//aza-Michael Isomerizations. J. Org. Chem. 2018, 83, 3516–3528. [Google Scholar] [CrossRef]
- Camp, J.E.; Greatrex, B.W. Levoglucosenone: Bio-Based Platform for Drug Discovery. Front. Chem. 2022, 10, 902239. [Google Scholar] [CrossRef]
- Diot-Néant, F.; Rastoder, E.; Miller, S.A.; Allais, F. Chemo-Enzymatic Synthesis and Free Radical Polymerization of Renewable Acrylate Monomers from Cellulose-Based Lactones. ACS Sustain. Chem. Eng. 2018, 6, 17284–17293. [Google Scholar] [CrossRef]
- Bonneau, G.; Peru, A.A.M.; Flourat, A.L.; Allais, F. Organic solvent- and catalyst-free Baeyer–Villiger oxidation of levoglucosenone and dihydrolevoglucosenone (Cyrene®): A sustainable route to (S)-γ-hydroxymethyl-α,β-butenolide and (S)-γ-hydroxymethyl-γ-butyrolactone. Green Chem. 2018, 20, 2455–2458. [Google Scholar] [CrossRef]
- Lefebvre, C.; Van Gysel, T.; Michelin, C.; Rousset, E.; Djiré, D.; Allais, F.; Hoffmann, N. Photocatalytic Radical Addition to Levoglucosenone. Eur. J. Org. Chem. 2022, 2022, e202101298. [Google Scholar] [CrossRef]
- Raviola, C.; Protti, S.; Ravelli, D.; Fagnoni, M. Photogenerated acyl/alkoxycarbonyl/carbamoyl radicals for sustainable synthesis. Green Chem. 2019, 21, 748–764. [Google Scholar] [CrossRef]
- Taniellan, C. Decatungstate photocatalysis. Coord. Chem. Rev. 1998, 178–180, 1165–1181. [Google Scholar] [CrossRef]
- Yamada, K.; Fukuyama, T.; Fujii, S.; Ravelli, D.; Fagnoni, M.; Ryu, I. Cooperative Polar/Steric Strategy in Achieving Site-Selective Photocatalyzed C(sp3)−H Functionalization. Chem. Eur. J. 2017, 23, 8615–8618. [Google Scholar] [CrossRef]
- Ravelli, D.; Fagnoni, M.; Fukuyama, T.; Nishikawa, T.; Ryu, I. Site-Selective C–H Functionalization by Decatungstate Anion Photocatalysis: Synergistic Control by Polar and Steric Effects Expands the Reaction Scope. ACS Catal. 2018, 8, 701–713. [Google Scholar] [CrossRef][Green Version]
- Mika, L.T.; Cséfalvay, E. Conversion of Carbohydrates to Chemicals. In Avanced Green Chemistry, Part 1; Horváth, I.T., Malacria, M., Eds.; World Scientific: Singapore, 2020; pp. 19–76. [Google Scholar]
- Zhu, J.; Yin, G. Catalytic Transformation of the Furfural Platform into Bifunctionalized Monomers for Polymer Synthesis. ACS Catal. 2021, 11, 10058–10083. [Google Scholar] [CrossRef]
- Martel, F.; Estrine, B.; Plantier-Royon, R.; Hoffmann, N.; Portella, C. Development of Agriculture Left-Overs: Fine Organic Chemicals from Wheat Hemicellulose-Derived Pentoses. Top. Curr. Chem. 2010, 294, 79–115. [Google Scholar]
- Bergman, J.A.; Kessler, M.R. Monomers and Resulting Polymers from Biomass. In Introduction to Chemicals from Biomass, 2nd ed.; Clark, J., Deswarte, F., Eds.; Wiley: Chichester, UK, 2015; pp. 157–204. [Google Scholar]
- Yue, X.; Queneau, Y. 5-Hydroxymethylfurfural Chemistry toward Biobased Surfactants. ChemSusChem 2022, 15, e202102660. [Google Scholar] [CrossRef]
- Velty, A.; Iborra, S.; Corma, A. Synthetic Routes for Designing Furanic and Non Furanic Biobased Surfactants from 5-Hydroxymethylfurfural. ChemSusChem 2022, 15, e202200181. [Google Scholar] [CrossRef]
- Girka, Q.; Hausser, N.; Estrine, B.; Hoffmann, N.; Le Bras, J.; Marinković, S.; Muzart, J. β-Amino acid derived gemini surfactants from diformylfuran (DFF) with particularly low critical micelle concentration (CMC). Green Chem. 2017, 19, 4074–4079. [Google Scholar] [CrossRef]
- Montagnon, T.; Kalaitzakis, D.; Sofadis, M.; Vassilikogiannakis, G. Chemoselective photooxygenations of furans bearing unprotected amines: Their use in alkaloid synthesis. Org. Biomol. Chem. 2016, 14, 8636–8640.
- Gollnick, K.; Griesbeck, A. Singlet oxygen photooxygenation of furans: Isolation and reactions of (4+2)-cycloaddition products (unsaturated sec.-ozonides). Tetrahedron 1985, 41, 2057–2068.
- Minaev, B.F. Electronic mechanisms of activation of molecular oxygen. Russ. Chem. Rev. 2007, 76, 988–1010.
- Schweitzer, C.; Schmidt, R. Physical Mechanisms of Generation and Deactivation of Singlet Oxygen. Chem. Rev. 2003, 103, 1685–1757.
- Schenck, G.O. Photochemische Reaktionen II. Über die unsensibilisierte und photosensibilisierte Autoxydation von Furanen. Justus Liebigs Ann. Chem. 1953, 584, 156–176.
- Cottier, L.; Descotes, G.; Nigay, H.; Parron, J.-C.; Grégoire, V. Photo-oxygénation des dérivés de l’hydroxyméthyl-5 furfural-2. Bull. Soc. Chim. Fr. 1986, 5, 844–850.
- Chauhan, D.K.; Battula, V.R.; Giri, A.; Patra, A.; Kailasam, K. Photocatalytic valorization of furfural to value-added chemicals via mesoporous carbon nitride: A possibility through a metal-free pathway. Catal. Sci. Technol. 2022, 12, 144–153.
- Desvals, A.; Fortino, M.; Lefebvre, C.; Rogier, J.; Michelin, C.; Alioui, S.; Rousset, E.; Pedone, A.; Lemercier, G.; Hoffmann, N. Synthesis and characterization of polymethine dyes carrying thiobarbituric and carboxylic acid moieties. New J. Chem. 2022, 46, 8971–8980
- Miles, W.H. Synthetic Applications of γ-Hydroxybutenolides. Curr. Org. Synth. 2014, 11, 244–267.
- Lefebvre, C.; Hoffmann, N. Les colorants et la lumière pour transformer la matière. Actual. Chim. 2019, 444–445, 38–43.
- Martel, J.; Tessier, J.; Demoute, J.-P. (Roussel Uclaf), Procédé de préparation de la 6,6-diméthyl-4-hydroxy-3-oxabicyclo(3.1.0)hexan-2-one et de ses éthers sous toutes leurs formes stéréoisomères. Eur. Pat. 1981, 0023454. [Google Scholar]
- Moradei, O.M.; Paquette, L.A. (5S)-(d-Menthyloxy)-2(5H)-furanone. Org. Synth. 2003, 80, 66–73. [Google Scholar]
- Marinković, S.; Brulé, C.; Hoffmann, N.; Prost, E.; Nuzillard, J.-M.; Bulach, V. Origin of Chiral Induction in Radical Reactions with the Diastereoisomers (5R)- and (5S)-5-l-Menthyloxyfuran-2[5H]-one. J. Org. Chem. 2004, 69, 1646–1651.
- Feringa, B.L.; de Jong, J.C. New Strategies in Asymmetric Synthesis Based On γ-Alkoxybutenolides. Bull. Soc. Chim. Belg. 1992, 101, 627–640.
- Gassama, A.; Ernenwein, C.; Youssef, A.; Agach, M.; Riguet, E.; Marinković, S.; Hoffmann, N. Sulfonated surfactants obtained from furfural. Green Chem. 2013, 15, 1558–1566.
- Gassama, A.; Ernenwein, C.; Hoffmann, N. Photochemical Key Steps in the Synthesis of Surfactants from Furfural-Derived Intermediates. ChemSusChem 2009, 2, 1130–1137.
- Hoffmann, N. Efficient photochemical electron transfer sensitization of homogeneous organic reactions. J. Photochem. Photobiol. C 2008, 9, 43–60.
- Tan, J.-N.; Ahmar, M.; Queneau, Y. Isomaltulose Oxidation and Dehydration Products as Starting Materials towards Fine Chemicals. Curr. Org. Chem. 2014, 18, 1768–1787.
- Tan, J.-N.; Ahmar, M.; Queneau, Y. Glucosyloxymethylfurfural (GMF): A creative renewable scaffold towards bioinspired architectures. Pure Appl. Chem. 2015, 87, 827–839.
- Tadiparthi, K.; Venkatesh, S. Synthetic approaches toward butenolide-containing natural products. Dihydro-5-(hydroxymethyl)-2(3H)-furanone. J. Heterocycl. Chem. 2022, 59, 1285–1307.
- Flourat, A.L.; Haudrechy, A.; Allais, F.; Renault, J.-H. (S)-γ-Hydroxymethyl-α,β-butenolide, a Valuable Chiral Synthon: Syntheses, Reactivity, and Applications. Org. Process Res. Dev. 2020, 24, 615–636.

