11. Introduction
The chemical transformation of biomass as a renewable feedstock as well as catalysis play a key role in sustainable chemistry
[1[1]]. In many cases, biomass is also an important alternative to fossils in the context of international economic and political dependencies and conflicts. In the context of chemical valorization, it should be pointed out that the molecular structure of biomass is different form corresponding fossil carbon compounds
[2][3][4[2,3,4]]. For example, carbohydrates which represent 75% of the biomass have a higher oxygen contain
[5][6[5,6]]. Also, lignin (about 20% of the biomass), which is the essential biomass-based source for aromatic compounds, contains higher amounts of oxygen
[7[7]]. The presence of numerous functional groups in such compounds makes them particular interesting for the production of new platform chemicals
[8][9][10][11][12[8,9,10,11,12]] for industry and also for many applications to fine chemistry and organic synthesis
[13][14][15][16][17][18][19][20[13,14,15,16,17,18,19,20]]. Catalysis is a further key element of green chemistry
[21[21]]. The catalytic transformation of biomass or biomass-derived platform or fine chemicals thus permits synergistic effects of sustainable chemistry as two requirements of green chemistry
[1[1]] are combined
[22][23[22,23]]. Figure 1 provides a graphical overview of the topic discussed herein.

Figure 1. Graphical overview.
Photochemical reactions have been recognized as being at the origin of sustainable chemistry
[24][25][26][27][28[24,25,26,27,28]]. They also play an important role in organic synthesis
[29][30][31][32[29,30,31,32]]. In this context, photochemical reactions also became very interesting in the chemical or pharmaceutical industry
[33[33]]. Applications of photochemical reactions in heterocyclic chemistry are particularly interesting
[34][35][36[34,35,36]]. Heterocycles are the partial structure of numerous bioactive compounds and play an important role in material science, for example for organic semi-conducting materials. More recently, photocatalytic reactions have gained an enormous interest in organic synthesis in the academic domain as well as in industry
[33][37[33,37]]. Various kinds of photocatalysis are reported
[38[38]]. They are characterized by different elementary steps such as sensitization by energy transfer, electron transfer or hydrogen transfer. The latter are often coupled and described as proton-coupled electron transfer (PCET)
[39][40][41[39,40,41]]. In particular, photocatalysis with visible light is now investigated
[42][43[42,43]]. The systematic study of these reactions opens perspectives for organic synthesis
[44][45[44,45]]. The generally mild photochemical reaction conditions facilitate a large number of C-H activation processes. Additionally, classical thermal reactions such as the Diels–Alder reaction can be facilitated
[46[46]]. Often in these cases, the reaction mechanism changes. A further important aspect is the combination of photoredox with other kinds of catalysis
[47][48][49][50][51][52[47,48,49,50,51,52]] such as enzyme
[53][54][55][56][57[53,54,55,56,57]], organocatalysis
[58][59][60][61][62[58,59,60,61,62]] or with conventional organometallic catalysis
[63][64][65][66][67][68[63,64,65,66,67,68]]. All these forms of catalysis are currently applied to the transformation of biomass or biomass-derived platform chemicals.
2. Photocatalytic Transformations of Biopolymers
By far most of the biomass is composed of polymers. Polysaccharides and lignins are the most important compound families. In many cases, photocatalytic transformation of such material yields mixtures of monomers
[9][69][70][71[9,69,72,73]]. For example, the depolymerization of cellulose is photo-catalyzed by TiO
2 [72][73][74[71,74,75]]. Reactive radical species are generated, which further react with oxygen. Under acidic conditions, such a catalysis may support the formation of furan derivatives, which are important platform chemicals. In a similar way, the photochemically supported Fenton oxidation has been used for depolymerization of polysaccharides
[75[76]].
Similar photoredox catalytic reactions have been carried out with lignin
[76][77][78][79[77,78,79,80]]. Lignin is an important renewable source for aromatic compounds
[7][80[7,81]]. For example, production processes of vanillin from lignin are frequently studied
[81[82]]. Another research approach with these material deals with the study of photochemical reactions
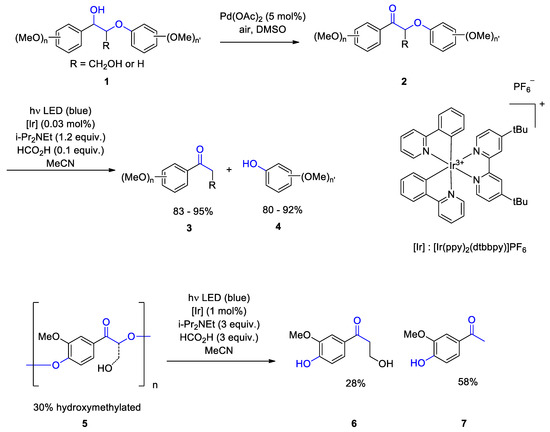
[82[83]]. Studies on the depolymerization with processed or native lignin are particular challenging. In order to obtain information on possibly efficient elementary steps, reactions of partial structures of lignin are investigated. Using Pd catalysis lignin fragments such as
1 are selectively oxidized to the corresponding acetophenone derivatives
2 (Scheme 1)
[83[84]]. Primary alkyl alcohol functions are not transformed under these conditions
[84[85]]. In a second photoredox catalytic reaction, C-O bond cleavage takes place yielding the acetophenone derivatives
3 and phenols
4. The iridium complex [Ir(ppy)
2(dtbbpy)]PF
6 was used as photoredox catalyst. The first process was separately performed and a workup without purification was carried out. This was necessary because different solvents were used in both steps. The resulting raw material was subjected to the photoredox catalytic step. The photoredox catalytic step was also carried out under similar conditions with model oligomers or polymers of lignin such as
5. The corresponding monomers
6 and
7 were isolated in high yields.
Scheme 1. Catalytic cleavage of lignin model dimers and polymers involving photoredox catalysis with visible light
Catalytic cleavage of lignin model dimers and polymers involving photoredox catalysis with visible light.
.
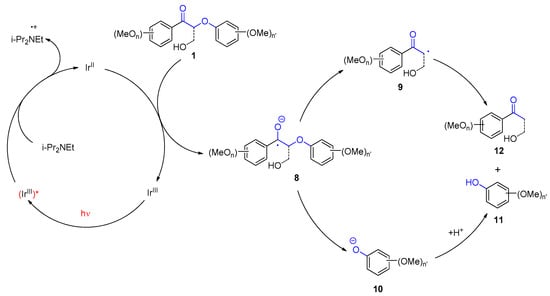
The following mechanism has been discussed for the photoredox catalytic part (Scheme 2). After excitation of the [Ir(ppy)
2(dtbbpy)] photocatalyst, which is an Ir
III species, electron transfer from the sacrificial amine donor occurs and an Ir
II species is generated. The latter is capable of reducing the acetophenone derivative
1, which leads to a radical anion
8. Fragmentation yields a neutral radical
9 and a phenolat
10. Protonation yields the final product
11. The neutral electrophilic radical undergoes hydrogen abstraction of hydrogen atom transfer (HAT), yielding the final acetophenone derivative
12. This step may also involve electron transfer followed by proton transfer
[85[86]]. Two catalytic processes are performed consecutively in the present transformation.
Scheme 2. Mechanism of the photoredox catalytic cleavage of lignin model dimers
Mechanism of the photoredox catalytic cleavage of lignin model dimers.
.
This transformation was also carried out with sunlight. Similar transformations of small model compounds have been reported
[86][87][88[87,88,89]]. Such studies have also been performed in the context of the sustainable production of vanillin
[89][90[90,91]]. In the context of organic photochemical reactions, for example cycloaddition with alkenes, derivatives of this compound are particularly interesting because in such transformations a high degree of molecular complexity and diversity is generated without chemical activation and thus reducing waste production
[27][91][92][93[27,92,93,94]].
3. Carbohydrates
Carbohydrates are versatile platform compounds and synthons in organic synthesis. As enantiomerically pure compounds they or their derivatives are key intermediates in asymmetric synthesis
[94][95[95,96]]. The systematic arrangement of chiral centers is particularly interesting in this regard
[96][97][98][99[97,98,99,100]]. Photocatalytic reactions have been applied in this domain
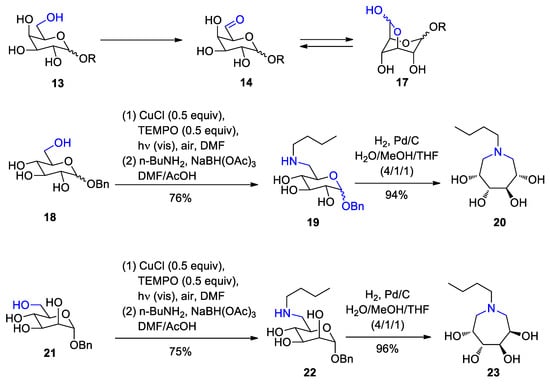
[100[101]]. In the present example, the selective oxidation of the primary alcohol function (
13) into an aldehyde (
14) plays a key role (Scheme 3)
[101[102]]. No secondary functions should be oxidized. Thus many syntheses with carbohydrate synthons can be facilitated since the number of protecting and deprotecting steps is reduced. The reaction is well known and can be carried out with galactose oxidase and molecular oxygen
[102[103]]. Since this enzyme activity is limited to galactose derivatives, various biomimetic strategies have been developed in order to extend the reactivity to further hexoses such as glucose or mannose derivatives
[103][104][105][106[104,105,106,107]]. Aside other methods, the Semmelhack reaction
[107[108]] can be used for the oxidation of primary alcohols to aldehydes
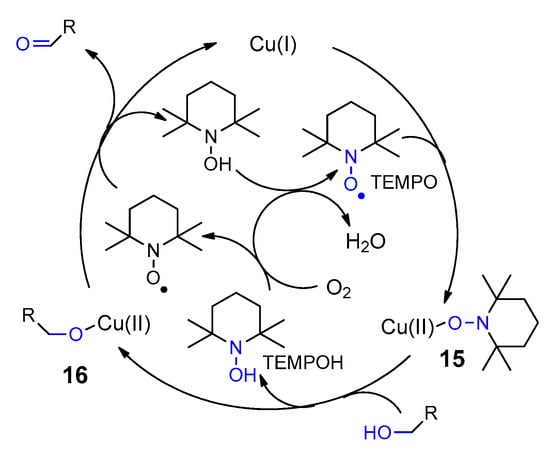
[108[109]]. Molecular oxygen is used as oxidant and copper salts and TEMPO (2,2,6,6-Tetramethylpiperidinyloxyl) are used as catalysts or as co-catalysts. Cu(II) acts as an oxidant (Scheme 4)
[109[110]]. This species (
15) is generated by the addition of TEMPO and release of TEMPOH. In this ligand exchange process, the primary alcohol is added (
16). The release of the carbonyl product regenerates the Cu(I) catalyst. The co-catalyst TEMPO is regenerated by the oxidation of TEMPOH by molecular oxygen. It has been observed that when the transformation is carried out with visible light irradiation, the reaction becomes efficient and can be applied to organic synthesis (Scheme 3)
[101[102]]. This effect can be explained by the fact that the Cu-O bond is weakened when such complexes are photochemically excited via a ligand to metal charge transfer (LMCT). Thus, ligand exchange steps in the mechanism are accelerated
[110[111]]. The oxidation product is stabilized by the formation of an anhydro derivative such as
17. Isolation and purification of these products were difficult. However, in situ transformation by a reductive amination efficiently yielded the corresponding glucose and mannose derivatives
19 and
22. Elimination of the benzylic protecting group followed by an intramolecular reductive amination yields the tetrahydroxyazepanes
20 and
23. Using the enzyme catalytic oxidation with galactose oxidase, tetrahydroxyazempanes with glucose- or mannose-related configurations are not accessible. Tetrahydroxyazepanes are azasugars. Such compounds possess various biological activities such as the inhibition of glycosidases and glycosyltranferases or anti-cancer activities
[111][112][113[112,113,114]]. In the present synthesis many steps of protecting and deprotecting different hydroxyfunctions and the utilization of corresponding reagents are avoided. As the irradiation was performed with visible light with visible light, the use of sunlight can be projected
[28][114[28,115]].
Scheme 3. Convenient synthesis of tetrahydroxyazepanes form glucose and mannose derivatives
Convenient synthesis of tetrahydroxyazepanes form glucose and mannose derivatives.
.
Scheme 4. Mechanism of the Semmelhack reaction
Mechanism of the Semmelhack reaction.
.
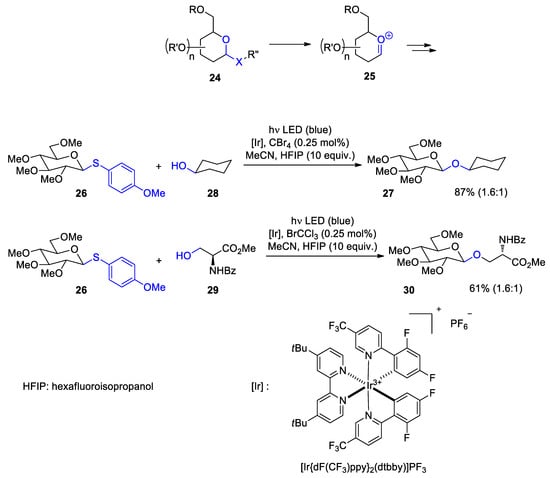
Glycosylation plays a central role in carbohydrate chemistry. During recent years various photocatalytic methods have been developed for this purpose
[115][116[116,117]]. Many of these reactions are carried out with visible light and with corresponding LED’s as light source. Such reaction conditions are particularly mild and energy consumption is low. Carboxonium intermediates
25 play an essential role in such transformations (Scheme 5)
[117][118[118,119]]. These species are generated from precursors
24 with corresponding leaving groups. Glycosylation then occurs by a nucleophilic attack. As these intermediates carry a positive charge, protonating agents or Lewis acidic compounds are often used as promotors. Oxidation such as photochemical electron transfer induced release of the leaving group is an alternative
[119[120]]. In this context, photoredox catalytic reactions have been systematically studied. Thus, compound
26 carrying an electron rich thioanisyl as the leaving group at the anomeric center was efficiently transformed with
28 into the corresponding glycosylation product
27 (Scheme 5)
[120[121]]. Additionally, more complex compounds such as the serine derivative
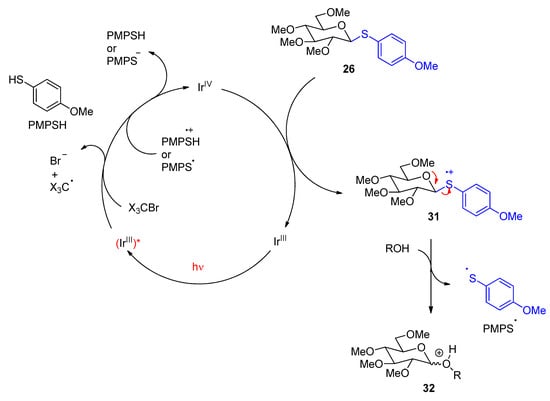
29 were successfully transformed. In most cases, the formation of β-anomers was favored. Tetrahalogenmethanes were added to the reaction mixture. They play a key role in the initiation step of the catalytic cycle. The following mechanism has been discussed. The reaction starts with the photochemical excitation of the iridium photocatalyst (Scheme 6). The Ir
III species is oxidized by a tetrahalogen methane. The resulting Ir
IV complex is reduced by the anisylthioether function (PMPS) in
26. The photocatalyst is thus regenerated. After the release of the PMPS radical from
31 and the nucleophilic, the attack of the alcohol compound takes place (
32). The photochemically excited Ir
III complex can also be oxidized in the reaction with intermediates of the PMPS leaving group. As it has already been pointed out, acidic activation of a leaving group leads to the formation of the carboxonium ion intermediate
25 (Scheme 5). Electronic excitation can significantly increase the acidity
[121][122[122,123]] and thus induce the release of a leaving group
[123[124]]. Thus, the trichloroimidate group was activated by photochemically excited phenols or naphthols
[124[125]]. Thiourea derivatives have also been used for the same purpose
[125[126]]. In some cases, both electron and proton transfer processes are involved in this activation
[126[127]].
Scheme 5. Photoredox catalytic glycosidation with anisylthioether derivatives. β-Anomeric glucosilation products are preferentially formed
Photoredox catalytic glycosidation with anisylthioether derivatives. β-Anomeric glucosilation products are preferentially formed.
.
Scheme 6. Mechanism of the Photoredox catalytic glycosidation with anisylthioether derivatives involving photochemical electron transfer with an iridium complex.
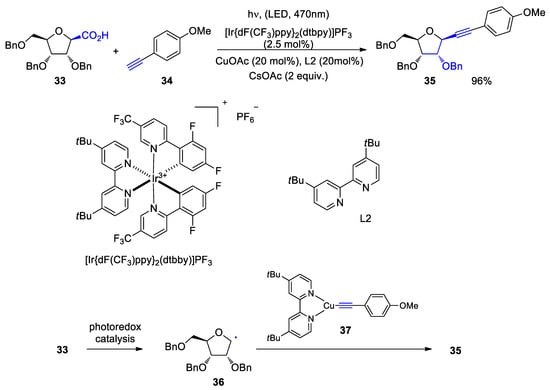
C-Glycosids play an important role as bioactive compounds
[127[128]]. In such compounds, an acetal C-O bond is replaced by a more stable C-C bond. Thus, many of these products possess enzyme inhibitor properties. For example, glucosidases or many nucleoside transformations can be inhibited. In the latter case, a C-N bond is replaced by a C-C bond
[128[129]]. In the reaction of the 1-β-d-glucofruanose derivative
33 with
p-methoxyphenylacetylene
34, only one diastereoisomer of the C-glucosid
35 was formed (Scheme 7)
[129[130]]. The photoredox catalytic part is carried out with an iridium catalyst. Compound
33 is decarboxylated and the intermediate
36 is formed. Such photodecarboxylation reactions are now frequently studied
[130[131]]. In the copper catalytic cycle, the complex
37 is formed, which reacts with the radical species
36 leading to the final product. The copper catalysis part is activated by addition of the ligand L2. In this case organic photocatalysts were less efficient. It cannot be completely excluded that the copper catalytic step is not affected by light absorption. In the present report, 28 C-glucosides have been synthesized with yields up to 96%. Heteroaromatic and alkyl acetylene derivatives have also been added to furanosyl radicals.
Scheme 7. Combined photo and copper catalysis applied to the coupling reactions of carbohydrate derived acids with alkynes
Combined photo and copper catalysis applied to the coupling reactions of carbohydrate derived acids with alkynes.
.
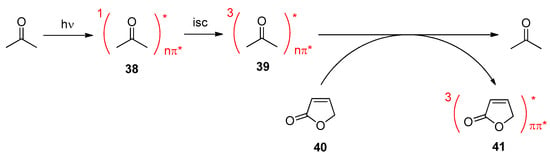
Chemical reactions can efficiently be induced by photosensitization. In this case, a chemical compound absorbs light and transfers its excitation energy to the substrate, which is electronically excited in this way. The latter then undergoes a photochemical reaction. The ground state of photosensitizer is regenerated and a new sensitizing process may take place. The photosensitizer acts as catalyst
[38[38]]. Often ketones are used as photosensitizer that efficiently transfer their triplet energies to a substrate. For example, this technique is applied to the photochemical transformation of α,β-unsaturated lactones. Often, these compounds possess a high excitation energy and low wavelength light irradiation is necessary for direct excitation to the S
1 state. Such conditions leads to unselective reactions. Since triplet energies are lower, triplet energy transfer from a sensitizer often leads to more selective reactions. More convenient reaction conditions such as room temperature instead of low temperature can be applied. Such a sensitization of a α,β-unsaturated butyrolactone or furanone is depicted in Scheme 8. Acetone absorbs light and is transferred to its singlet state S
1 38 possessing nπ* character. After intersystem crossing to the corresponding energy lower triplet state, T
1 39 is reached. Triplet energy transfer now occurs to the furanone
40 that is thus transferred to its triplet state T
1 41 with ππ* character. The latter species undergoes photochemical reactions. Acetone also absorbs at lower wavelengths (λ
max (nπ*) = 278 nm)
[131[132]]. However, irradiation at longer wavelengths such as 300 nm generates sufficient excited molecules for sensitization when acetone is used as a solvent or co-solvent.
Scheme 8. Electronic excitation of a furanone to the triplet state via triplet energy transfer
Electronic excitation of a furanone to the triplet state via triplet energy transfer.
.
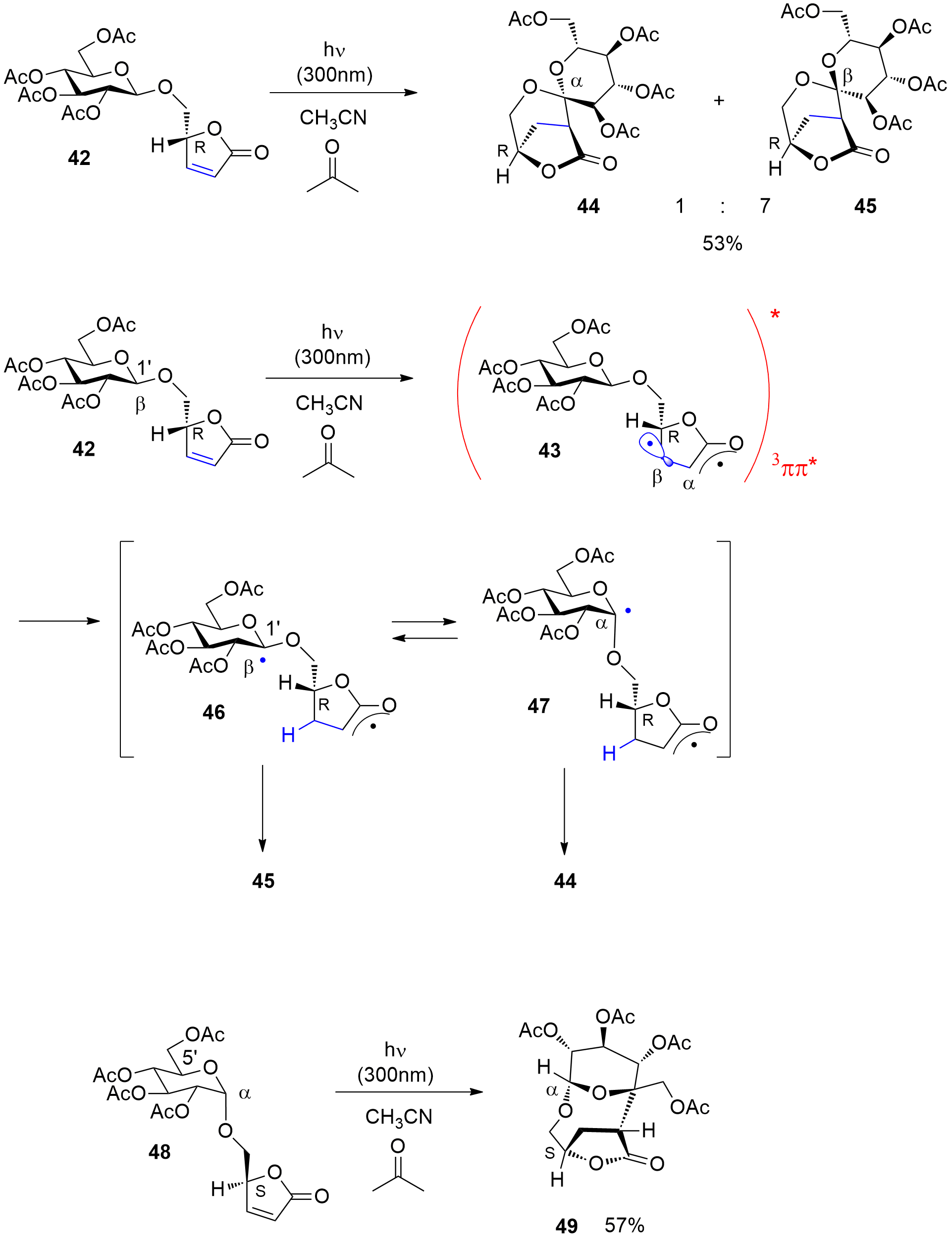
Under these conditions, furanone compounds such as
42 carrying a glucosyl moiety have been electronically excited to their triplet state
43 (Scheme 9)
[132[133]]. Sugar derivatives with their secondary alcohol functions are excellent hydrogen donors for hydrogen abstraction or hydrogen atom transfer (HAT) processes
[133][134[134,135]]. Such reaction steps are also observed in non-catalytic photochemical transformations. For some examples, see refs.
[135][136][137[136,137,138]]. Hydrogen abstraction is observed with many triplet exited compounds
[93][134][136][138][139[94,135,137,139,140]]. In the photosensitized reaction of glucosylated furanone derivative
42, the spirocyclic compounds
44 and
45 are formed. In contrast to many conventional radical addition reactions of such α,β-unsaturated lactones, a C-C bond is formed in the α-position. The electronic excitation occurs via triplet energy transfer from acetone to
42. The
3(ππ)* state (
43) is characterized by the suppression of the C=C π-bond and an increased spin density in the β-position
[134][140[135,141]]. For this reason, hydrogen abstraction occurs from the anomeric center of the sugar moiety into this position and the intermediates
46 and
47 are formed, both in conformational equilibrium. Radical combination yields the final products
44 and
45. In this reaction, the regioselectivity of hydrogen abstraction depends on the relative configuration of the chiral centers in the anomeric position and the chiral center at the furanone moiety. When the corresponding α-anomer diastereoisomer
48 is transformed under the same conditions, hydrogen abstraction occurs at the position 5′ of the glucosyl substituent and the macrocyclic product
49 is formed. Similar reactions have been carried out with direct excitation of a furanone moiety to the singlet state and subsequent intersystem crossing to the triplet state. The transformation under these conditions needed lower wavelength irradiation and low reaction temperature
[141[142]]. It should further be pointed out that for the triplet sensitized of C-H activation, no particular reagents are needed, which often leads to the production of additional waste. Furthermore, acetone is used as a co-solvent and no chromatographic separation of the sensitizer or a photocatalyst is necessary.
Scheme 9. Intramolecular photosensitized radical addition of a carbohydrate moiety to a furanone. Acetone was used as sensitizer and cosolvent in a ratio of 1/4 with acetonitrile.
4. Fatty Acids
Fatty acids obtained from triglycerides play an important role in the chemical valorization of biomass. Their use for the production of surfactants belong to the basic culture techniques of humanity. More recently their application as part of biofuel is reported [142[143]]. Fatty acid methyl esters (FAME) can be obtained by photocatalytic esterification to produce biodiesel [143][144][145[144,145,146]].
Photodecarboxylation is an interesting transformation both for biofuel production and in organic synthesis [146][147][148[147,148,149]] as it leads to radical intermediates that can form alkanes or further react with a variety of substrates. Decarboxylation is also carried out using photochemical electron transfer [149][150][151][152[150,151,153,154]]. Such reaction steps also play an important role in many metal catalyzed reactions [130][149][153][154[131,150,155,156]]. In this context, but also for application to organic synthesis, decarboxylation is an interesting transformation as it leads to a variety of radical intermediates.
Hatanaka et al. have transformed palmitic acid into the corresponding n-pentadecane [155[157]]. This method consists of the photoelectron transfer of the carboxylate with a photogenerated cation radical of phenantrene followed by decarboxylation. The hydrogen abstraction from a thiol results in the formation of the corresponding alkane. Nicewicz et al. used of an acridinium photooxidant in the presence of an organic base for a decarboxylation [156[158]]. The mechanism involves a photochemical electron transfer to the acridinium from the carboxylate followed by CO2 release. The generated carbon radical abstracts a hydrogen from the in situ generated thiophenol. Wang et al. reported a route for photocatalytic decarboxylation for alkane production using fatty acids. Their system uses semiconductor Pt/TiO2 and irradiation using 365 nm LEDs, and a hydrogen atmosphere resulted to be crucial for the high yields (>90%), while several fatty acids were successfully transformed in the corresponding alkanes [157[159]].
Enzyme-catalyzed reactions play a key role in the transformation of biomass or biomass-derived compounds. Recently, an algal photoenzyme named fatty acid photodecarboxylase (CvFAP) was discovered. This photoenzyme transforms fatty acids into the corresponding alkanes upon light irradiation [158][159][160[160,161,162]]. Among the different uses of this photo-enzyme, several methods were reported to obtain, for example, bio-propane and bio-butane from fatty acids [161][162[163,164]], and also bio-diesel [163[165]] depending on the substrate that was used.
Concerning the application of such reactions to organic synthesis, radical intermediates are often generated by decarboxylation of carboxylic acids or their derivatives [164][165][166][167][168[166,167,168,169,170]]. These radicals undergo addition to various acceptor molecules. In many of these reactions, photocatalysis involving electron transfer is applied. The transformations are often also carried out with corresponding fatty acids or their derivatives.
5. Biomass-Derived Platform Chemicals
Levoglucosenone is an interesting platform chemical, which is obtained from pyrolysis of cellulose under acidic reaction conditions [169][170][171[177,178,179]]. Recently, it gained additional interest as a precursor of a biobased dipolar solvent [172[180]]. Levoglucosenone is now produced on the industrial scale. Different functional groups are localized on a relatively small enantiomerically pure molecule. Thus, it becomes a very attractive synthon for organic synthesis [173][174][175][176][177[181,182,183,184,185]]. Levoglucosenone can also be transformed into further platform chemicals such as furanone derivatives [178][179[186,187]].
Photocatalytic reactions have also been performed with this compound. Using tetra-n-butylammonium decatungstate (TBADT) as a photocatalyst, various radical species have been added to levoglucosenone 50 (Scheme 10) [180[188]]. Thus, alkanes (51a) or cyclic ethers (51b) have been added. The addition of formamide (51c) was particular efficient. The method was also used to the addition of aromatic (51d), heteroaromatic (51e) and aliphatic aldehydes (51f). The scope for the addition of aldehydes is large [181[189]]. From these precursors radical intermediates are generated by hydrogen atom transfer (HAT) to the photochemically excited TBADT (Scheme 11) [182][183][184[190,191,193]]. The radicals add stereospecifically anti with respect to the (‑CH2‑O‑) moiety in 50. The energy difference for both diastereotopic attacks of the radical intermediates is 5 kcal mol−1. The configuration of further chiral centers generated in the reaction as in 51b was not controlled. The electrophilic radical intermediate 52 is reduced by [HW10O32]4−, which leads to the final product 51. In the context of sustainable chemistry, it should further be mentioned that such reactions have been carried out with sunlight and under very simple apparatus conditions [183[191]].
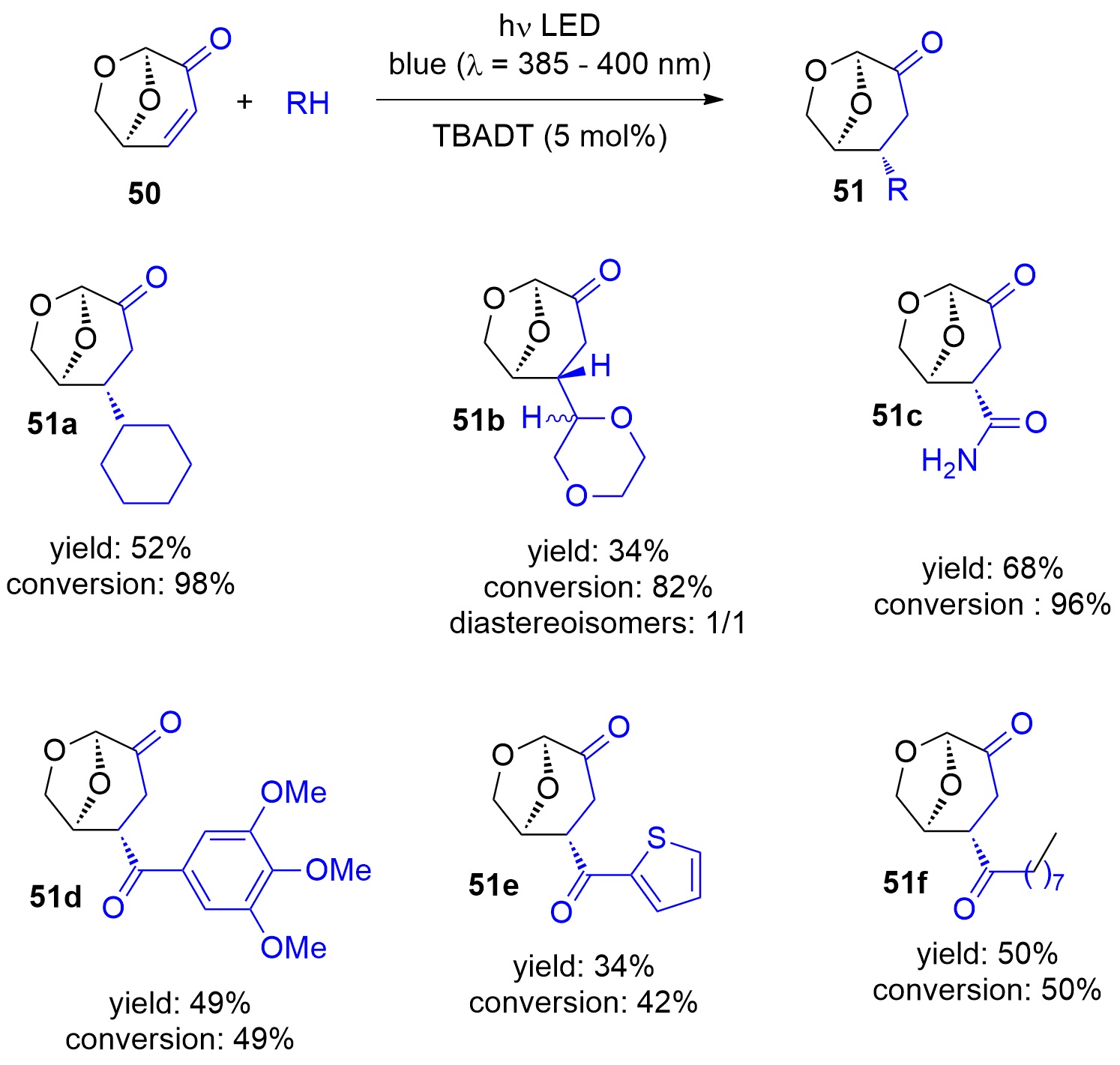
Scheme 10. Photocatalytic addition of radicals to levoglucosenone
Photocatalytic addition of radicals to levoglucosenone
50
.
.
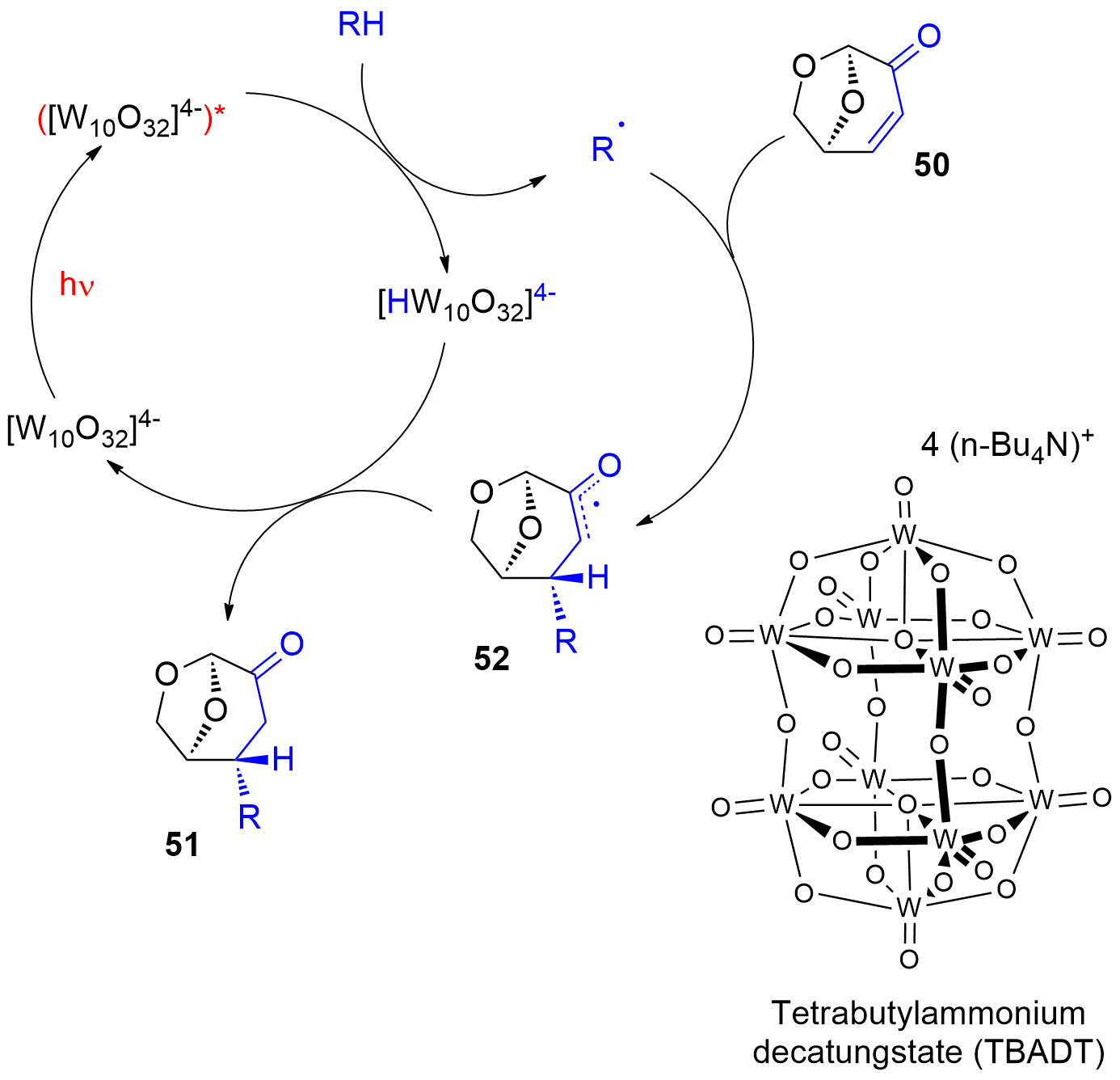
Scheme 11. Mechanism of the photocatalytic addition of radicals to levoglucosenone
Mechanism of the photocatalytic addition of radicals to levoglucosenone
50.
Furans are valuable platform chemicals obtained from carbohydrates [185][186][187[194,195,196]]. They are formed by the dehydration of sugars. Thus, dehydration of pentoses or hemicellososes yields furfural while corresponding transformations of hexoses lead to hydroxymethylfurfural (HMF). These compounds are currently studied in connection with the production of bulk products such as new polymers [185][188[194,197]] or surfactants [189][190][191[198,199,200]].
One of the most-used photocatalytic reactions of furans is the photooxygenation of these compounds involving singlet oxygen [192][193[229,230]]. Oxygen is one of the rather rare molecules possessing a triplet multiplicity at the ground state [194][195[201,202]]. The photooxygenation of furfural 53 with singlet oxygen yields hydroxyfuranone (Scheme 12) [196][197][198[231,232,233]]. In most cases of photooxygenation of furan derivative, an endoperoxide such as 54 is formed. As the reaction is carried out in an alcohol as solvent, this intermediate undergoes a nucleophilic attack of an alcohol molecule leading to the final products 5-hydroxyfuran-2[5H]-one 55 and a formic ester 56. This very convenient oxidation can be carried out in the laboratory at 100 g scale [199[234]]. Hydroxyfuranone 55 is itself a platform chemical with numerous applications to organic synthesis [200][201[235,236]]. Chiral synthons can be obtained from this compound and many efficient applications to asymmetric synthesis have been reported [202][203][204][205[237,238,239,240]]. Hydorxyfuranone 55 was also used for the preparation of biodegradable surfactants (Scheme 13) [206[241]]. A photoredox catalytic process was also applied for the preparation of such compounds [207][208[242,243]].
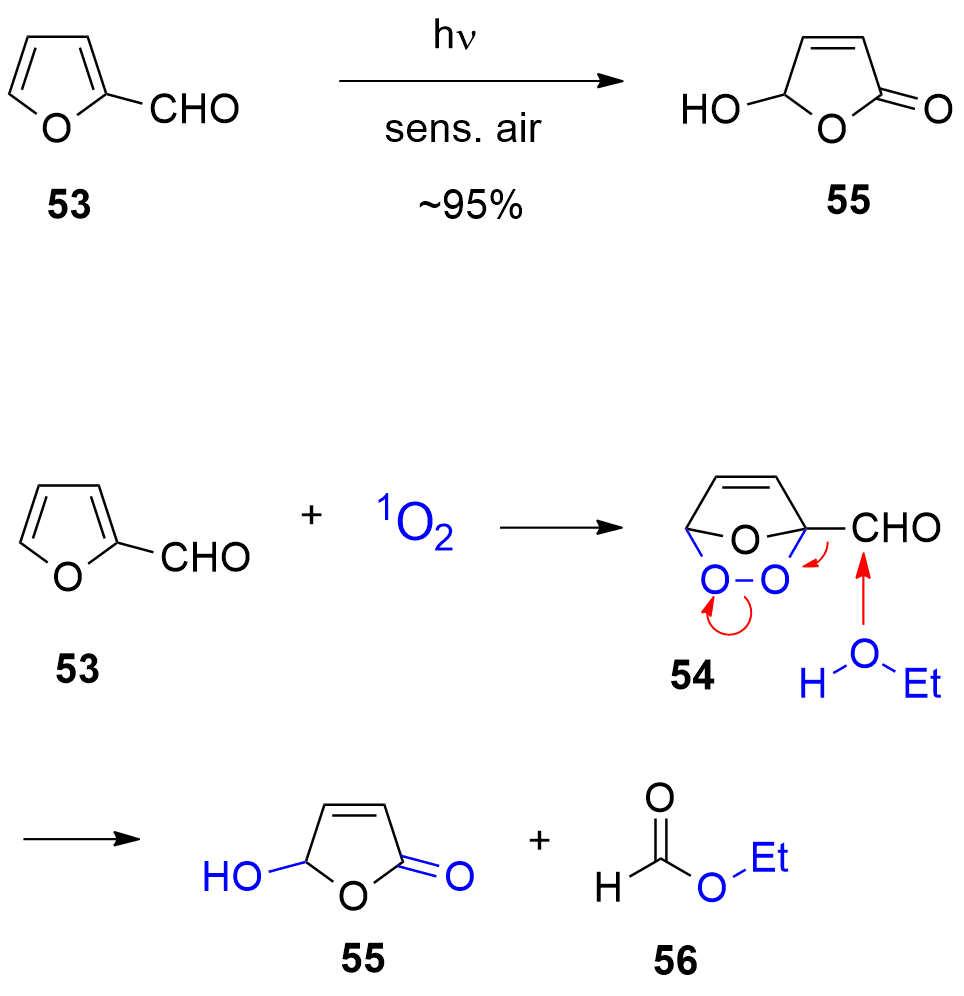
Scheme 12. Photooxygenation of furfural
Photooxygenation of furfural
53 to hydroxyfuranone
to hydroxyfuranone
55
.
.
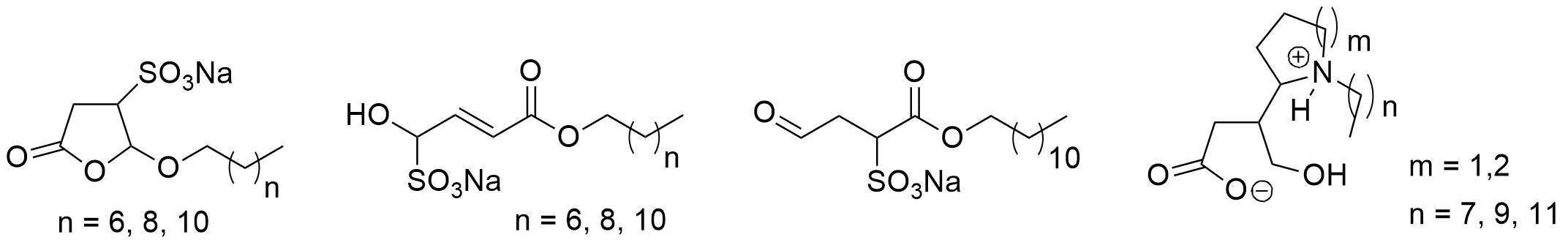
Scheme 13. Surfactants obtained from furfural
Surfactants obtained from furfural.
.
Photooxygenation was also applied as a key step to the synthesis compound 57 (Scheme 14, also compare Scheme 9). Using conventional carbohydrate chemistry methodology, it is often difficult to prepare α-anomeric derivatives of hexose pyranoses such as derived from glucose for example. Isomaltulose is a disaccharide obtained by enzymatic isomerization of saccharose [209][210[244,245]]. In the isomaltulose molecule, a fructose moiety in its furanose form is connected in the α-anomeric position of glucose. Selective dehydration on the fructose furanoside under acidic conditions leads to the formation a hydroxymethylfurfural derivative (58) [246]. Acylation (59) and photooxygenation yields the product 60. Finally, reduction of the hydroperoxide 60 leads to 57 [132[133]]. The present synthesis also provides a very convenient access to 5-hydroxymethylfuranones such as 57. Using conventional methods of carbohydrate chemistry, these compounds are prepared with multi-step syntheses from sugars or amino acids [211][212[247,248]].
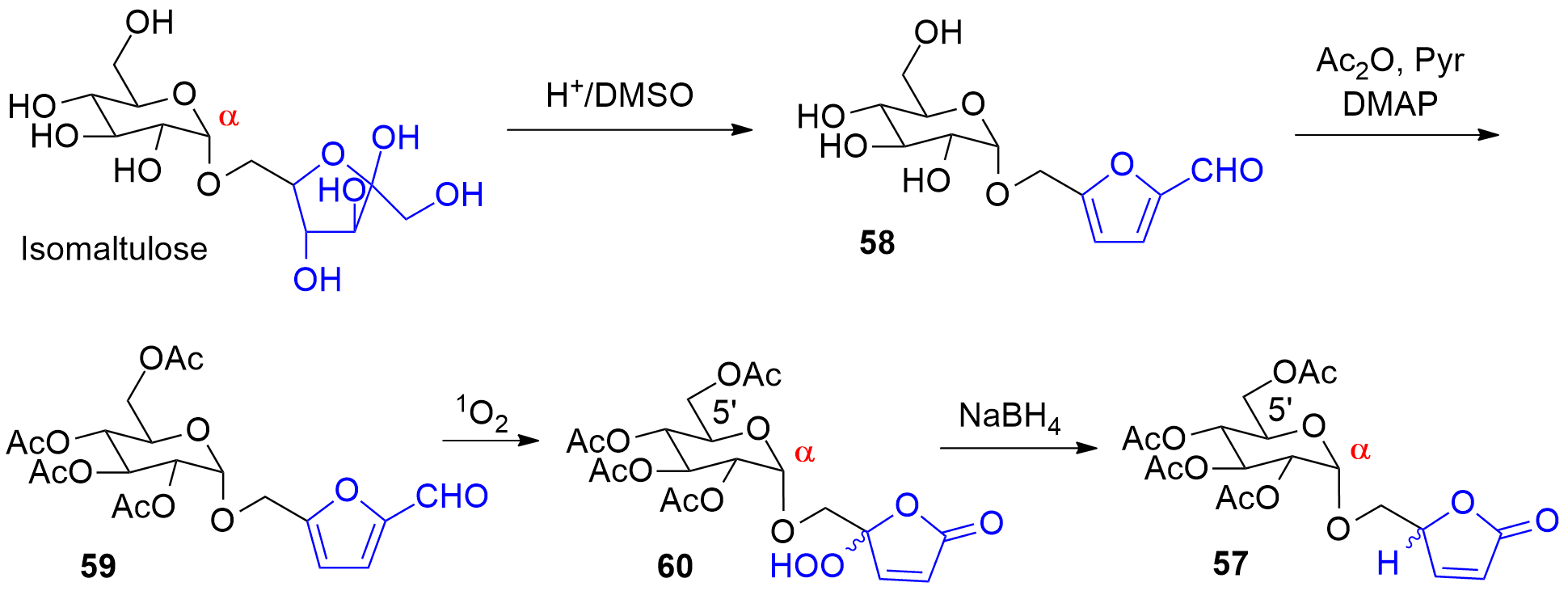
Scheme 14. Synthesis of a α-connected glycosyl furanone
Synthesis of a α-connected glycosyl furanone
57. from isomaltulose
. from isomaltulose.
.
6. Conclusion
Biomass or biomass-derived platform chemicals have become an alternative feedstock for chemical industry. They may replace conventional fossil-based resources in two senses. On one hand biomass based compounds can be used for the synthesis of already existing products in the chemical industry as they are available mainly from petroleum. On the other hand, these biomass or biomass-derived compounds can be used to produce new and original compounds, which is facilitated by characteristic structure elements such as the presence of numerous hydroxyl functions in carbohydrates. Photochemical or photocatalytic reactions make compounds or compound families available that cannot or difficultly synthesized using conventional methods of organic synthesis. A high degree of molecular complexity and diversity is thus generated in a facile way. This is explained by the complementarity of ground state and excited state reactivity. In this same context, it should be pointed out that in the past, photochemical or photocatalytic reactions of biomass-derived products have rarely been investigated in a systematic way.
Photochemical and catalytic reactions on one hand and the utilization of biomass as a renewable feedstock on the other are part of sustainable chemistry. The combination of both opens perspectives for sustainable chemistry.