Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shusei Mizushima | -- | 2102 | 2023-06-19 11:37:14 | | | |
| 2 | Rita Xu | Meta information modification | 2102 | 2023-06-19 11:55:35 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mizushima, S.; Sasanami, T.; Ono, T.; Kuroiwa, A. Intracytoplasmic Sperm Injection for Avian Genome Editing. Encyclopedia. Available online: https://encyclopedia.pub/entry/45783 (accessed on 24 June 2026).
Mizushima S, Sasanami T, Ono T, Kuroiwa A. Intracytoplasmic Sperm Injection for Avian Genome Editing. Encyclopedia. Available at: https://encyclopedia.pub/entry/45783. Accessed June 24, 2026.
Mizushima, Shusei, Tomohiro Sasanami, Tamao Ono, Asato Kuroiwa. "Intracytoplasmic Sperm Injection for Avian Genome Editing" Encyclopedia, https://encyclopedia.pub/entry/45783 (accessed June 24, 2026).
Mizushima, S., Sasanami, T., Ono, T., & Kuroiwa, A. (2023, June 19). Intracytoplasmic Sperm Injection for Avian Genome Editing. In Encyclopedia. https://encyclopedia.pub/entry/45783
Mizushima, Shusei, et al. "Intracytoplasmic Sperm Injection for Avian Genome Editing." Encyclopedia. Web. 19 June, 2023.
Copy Citation
Poultry are one of the most valuable resources for human society. They are also recognized as a powerful experimental animal for basic research on embryogenesis. Demands for the supply of low-allergen eggs and bioreactors have increased with the development of programmable genome editing technology. The CRISPR/Cas9 system has recently been used to produce transgenic animals and various animals in the agricultural industry and has also been successfully adopted for the modification of chicken and quail genomes.
CRISPR/Cas9
intracytoplasmic sperm injection
primordial germ cells
1. Introduction
Poultry, such as the chicken (Gallus domesticus), turkey (Meleagris gallopavo), and Japanese quail (Coturnix japonica), are commercially important species because they serve as major food sources worldwide. The poultry industry is indispensable for supporting sustainable development worldwide and is expected to significantly contribute to the United Nations’ Sustainable Development Goals. Studies on the embryos of these birds have contributed to a more detailed understanding of organogenesis [1][2] and human diseases [1], due to their oviparity. Chicken and quail embryos are easy to access and, thus, are amenable to introducing transgenes, normal or transgenic primordial germ cells (PGCs), and viruses [3][4]. The draft genome sequences of chicken, quail, turkey, and zebra finch genomes have been generated, with the information obtained supporting the generation of transgenic birds and the mass production of recombinant proteins [5][6][7][8]. The generation of gene knockout animals is a powerful approach for identifying essential proteins and the functions of uncharacterized genes in many species, as is the case in birds. The combination of recent technical advances is expected to provide novel insights into future aspects of avian biotechnology as well as human society.
The generation of gene knockout chickens by homologous recombination using PGC-mediated methods was initially reported in 2013 [9] following its establishment in mice [10]. Immunoglobulin light chain knockout chickens were subsequently generated using the same method [11]. Although these achievements contributed to the progression of avian gene targeting, the efficiency of this recombination was very low. In this regard, the emergence of site-specific nuclease has provided new avenues for modifications to avian genomes [12][13][14]. The earliest programmable genome editing tools for the generation of gene knockout animals were zinc finger nuclease (ZFN) and transcription activator-like effector nuclease (TALEN) [15]. These enzymes were artificially created by the fusion of FoKI endonucleases with the capacity to recognize long-chain DNA. FoKI endonucleases recognize target DNA and induce double-strand breaks (DSB) and small indels by error-prone non-homologous end joining (NHEJ).
2. CRISPR/Cas9-Mediated Genome Editing Technology
The CRISPR/Cas system, originally found in bacteria and archaea, is an RNA-based adaptive immune system that destroys invading plasmids, phages, and viruses [16][17][18]. The nucleoprotein complex, consisting of three crucial components: CRISPR-coding RNA (crRNA), trans-activating crRNA (tracrRNA), and Cas protein, recognize exogenous DNA and degrade it by endonuclease activity [19], causing error-prone NHEJ. The binding site of Cas9 is located upstream of the protospacer adjacent motif (PAM) containing the 5′-NGG base sequence. Humanized Cas9 protein or Cas9 derived from Streptococcus pyogenes Cas9 (SpCas9), combined with a synthetic single guide RNA (sgRNA) produced by fusing crRNA with tracrRNA, has been shown to trigger DSB in mammalian cells [20][21]. This CRISPR/Cas9 system may be easily designed and prepared for a plasmid vector by changing the sgRNA sequence to a specific region of the genome sequence instead of ZFN and TALEN described above, and its use has rapidly expanded. The recent re-engineering of Cas9 has resulted in the establishment of dead Cas9 (dCas9) with mutations in two nuclease domains, which has extended the application of the CRISPR/Cas9 system with the potential transcriptional inhibitor/activator, and point mutations [22][23][24][25] (Figure 1A).
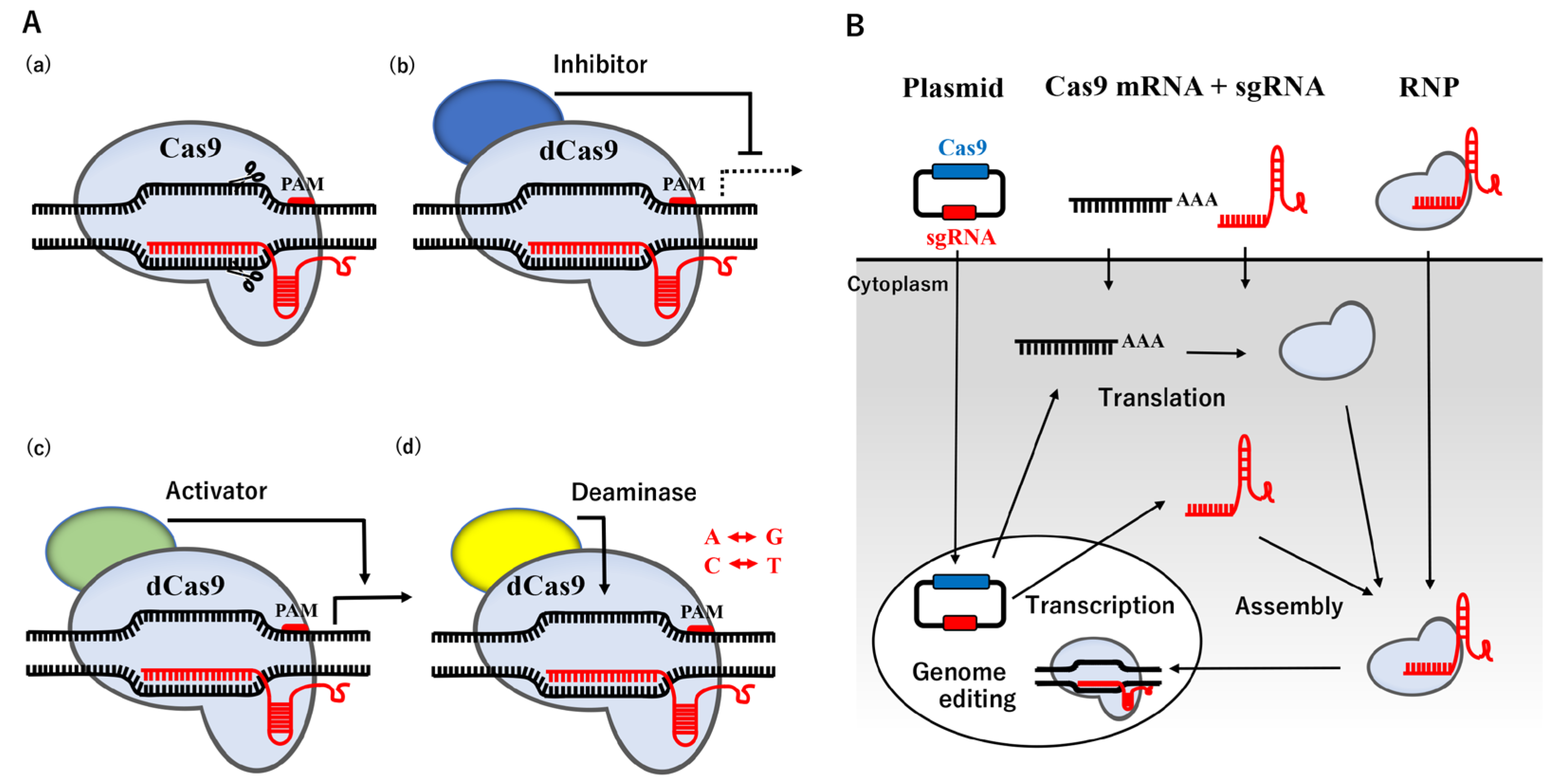
Figure 1. Applications and Routes of the CRISPR-Cas9 system. (A) Schematic illustration of the applications. (a) A basic molecular model of the CRISPR/Cas9 system. (b) dCas9 fused with a transcriptional inhibitor (blue) represses transcription. (c) dCas9 fused with a transcriptional activator (green) boosts transcription. (d) dCas9 bound to adenine or cytosine deaminase (yellow) modifies A to G or T to C, respectively. (B) Schematic illustration of the routes. Three forms of plasmids encoding Cas9 and sgRNA, the RNAs of Cas9 mRNA and sgRNA, or a complex of Cas9 protein and sgRNA are available for delivery into cells by transfection or a microinjection.
Three viral vectors have so far been adopted in clinical trials for the delivery of the CRISPR/Cas9 system: adeno-associated viruses (AAV), adenoviruses, and lentiviruses [26]. AAV are preferred due to their low immunogenicity and stable expression. However, the construction of long Cas9 sequences in plasmids is difficult due to the packaging limitation of AAV. This issue may be partially resolved using truncated SpCas9 [27]. On the other hand, the packaging capacities of adenoviruses and lentiviruses are high. In addition, lentiviruses and adenoviruses show high infection efficiencies in non-dividing cells and non-/dividing cells, respectively. However, lentivirus vectors generally induce insertion mutations through the sustained expression of Cas9 and sgRNA, which may result in off-target effects, while other adenoviruses may induce immune toxicities [28].
In a non-viral method, three forms of the CRISPR/Cas9 system are now available for the delivery of a nucleoprotein complex into the nucleus: plasmid DNA, the RNA system of Cas9 mRNA and sgRNA, and the RNA-protein complex of sgRNA and Cas9 ribonucleoprotein (Cas9 RNP) (Figure 1B). In the plasmid delivery system, the CRISPR/Cas9 plasmid, termed pX330, was originally constructed [20]. The pX330 plasmid contains two expression cassettes, with the expression of sgRNA being driven under the U6 promoter in one and that of Cas9 being driven under the chicken β-actin promotor in the other. In addition, Cas9 is engineered to carry the nuclear localization signal, similar to SV40, which is required to transport the CRISPR/Cas9 system into the nucleus [29]. The RNA system is capable of controllable release into the cytoplasm. The Cas9 RNP system may skip the expression of the Cas9 protein and sgRNA in cellular events, but it also reduces the risk of off-target effects because it avoids overexpression. Although a gene delivery system, such as electroporation and nanoparticle transfer, may be applied to almost any cell type at any stage of the cell cycle, even large-molecule particles, low delivery efficiency remains a challenge due to the large molecular size, instability, and the low efficiency of the endosomal escape [30][31].
3. Current Approaches for Avian Genome Editing Based on the CRISPR/Cas9 System
Avian genome editing technology is based on a procedure for establishing transgenic chicken lines. Various approaches have been attempted to create transgenic birds, including viral infection, the chimeric method, and sperm-mediated gene transfer (Figure 2).
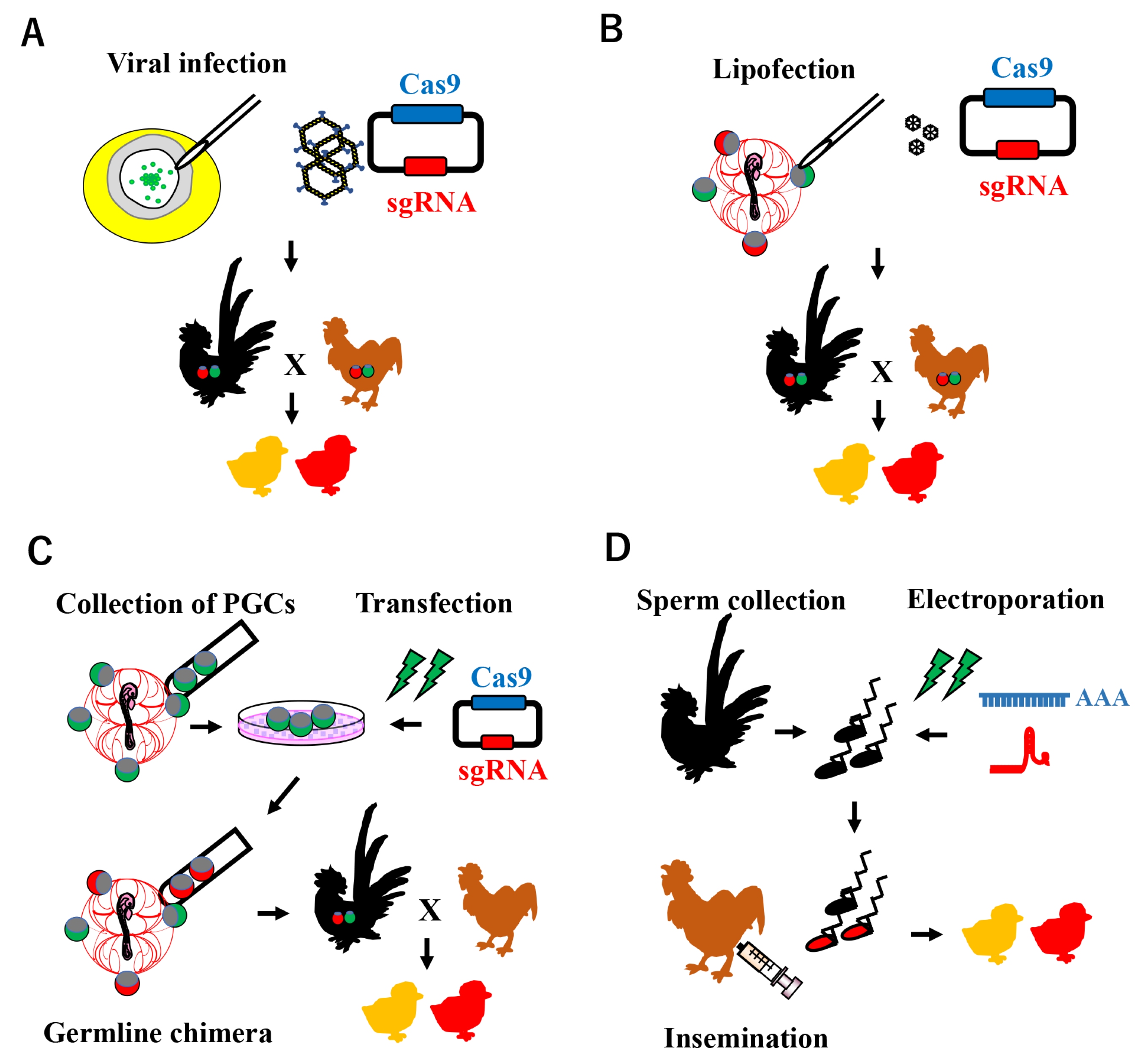
Figure 2. Schematic illustration of CRISPR-Cas9 system-mediated genome editing in poultry. (A) Viral infection model: a recombinant adenovirus carrying CRISPR/Cas9 components is infected at blastoderm stage X, where PGCs exist in the center area. (B) PGC-mediated genome editing: the plasmid-encoding Cas9 and sgRNA expression cassettes with lipofectamine are microinjected into embryonic blood vessels, in which the PGCs are circulating. (C) PGC-mediated genome editing: the CRISPR/Cas9 system is introduced into cultured PGCs in vitro, which are mainly collected from embryonic blood vessels. Enriched genome-edited PGCs are transferred into the blood vessels of recipient embryos to generate a germline chimera. (D) Sperm Transfection-Assisted Gene Editing (STAGE): a mixture of Cas9 mRNA and sgRNA is transfected into ejaculated sperm collected from roosters, which are then subjected to artificial insemination in hens.
3.1. Viral Infection
Viral infection is the most reliable method because of its high efficiency for incorporation of the transgene. The viral gene delivery system was primarily applied to blastoderm stage-X embryos for the purpose of inducing the transgene into the genome of germline cells in the blastoderm [32][33][34][35][36][37][38][39]. Salter et al. [32] was the first to attempt retrovirus transfection into a blastoderm. The infection of a replication-defective pantropic retrovirus vector based on Moloney murine leukemia virus (MoMLV) pseudotyped with the vesicular stomatitis virus G protein was subsequently attempted in Japanese quail stage-X embryos [33]. In that study, the viral vector sequence was detected in the tissue of all the hatched quails, and the efficiency of the germline transmission from G0 to G1 was very high (80%). In addition, the injection of the lentiviral vector derived from the lentivirus equine infectious anemia virus into stage-X embryos was successfully adopted to produce transgenic chickens [34]. Although the expression of the anti-prion single-chain Fv protein was successfully achieved in the eggs of transgenic chickens transfected with the MoMLV-based mouse stem cell virus, the expression level of this protein in the G2 generation decreased to less than that of the G0 founder due to a transgene silencing effect [35]. Therefore, the appropriate selection of key regulator regions for gene silencing or increases in transcriptional activity may be required in future studies that focus on the expression of reporter genes and analyze their functions.
The generation of genome-edited quails was initially achieved using an adenovirus injection containing the CRISPR/Cas9 system targeting the melanophilin (MLPH) gene [40] (Figure 2A). Since MLPH functions in feather pigmentation, the feathers of the genome-edited quails were gray. In this study, germline chimeric G0 quails produced genome-edited progeny, and the efficiency of the germline transmission ranged between 2.4 and 10%, suggesting the efficacy of adenovirus infection as a gene delivery system to transduce the CRISPR/Cas9 system for proliferating blastodermal cells. In addition, myostatin gene-edited quails with a higher body weight and muscle mass were successfully produced using the same adenovirus injection [41]. The effects of the adenoviral transduction of the CRISPR/Cas9 system into local tissue were also demonstrated in chick leg muscle [42]. More recently, genome-edited ducklings were successfully produced by an adenoviral infection into blastoderms, indicating the efficacy of genome editing technology with adenoviruses in poultry and water birds other than chickens and quails [43].
3.2. Chimeric Method Using PGCs
Since the viral method may introduce a potentially hazardous infection, non-viral methods for the integration of exogenous DNA into the host genome are preferred. Germ cells are the sole source of the transmission of genetic information to the next generation; therefore, the use of PGCs is currently the most common method for cell-mediated gene transfer because of the higher competency of PGCs than other stem cells, such as embryonic stem cells and embryonic germ cells [44][45][46]. Chicken PGCs appear from the early blastoderm stage and are present as colonies in the central region of the blastoderm at stage X [47][48][49]. After migrating to the germinal crescent region, they circulate in embryonic blood vessels and settle in the embryonic gonads [50]. Due to the unique migratory features of avian PGCs, they may be isolated from various embryonic stages and subjected to long-term cultures without the loss of germ cell potency [51]. Wentworth et al. [52] reported that cultured PGCs settled in recipient gonads after their injection into embryonic blood vessels, resulting in the production of a germ-line chimera in chickens. Isolated gonadal PGCs retained the capacity to migrate and differentiate into mature gametes in the recipient embryo transplanted into their blood vessels [53][54][55].
Using the PGC transplantation method, van de Lavoir et al. [51] reported the successful expression of green fluorescent protein, the reporter gene, in germline chimeras. The transposon system has since been identified as the most efficient for transgenesis [56][57]. Transposon elements have the ability to change their positions within a genome, which mediates the integration of the intended foreign gene. Park and Han reported about 50% germline chimerisms using piggyBac and the Tol2 transposon [57]. Another method involved the direct injection of transposon plasmids into embryonic blood vessels with lipofectamine, with the transformation of the circulating PGCs and the subsequent production of transgenic chickens [58] (Figure 2B). Zhu et al. successfully integrated human immunoglobulin-coded genes in embryonic stem cells derived from stage X [59], indicating the utility of transgenic poultry in industries such as agriculture and biomedicine.
Genome-editing technologies using the PGC method have been developed in the last decade for basic research on embryogenesis as well as for agriculture and biomedicine. In 2016, two egg-white genes, ovalbumin and ovomucoid, which are well-known allergenic proteins, were efficiently mutagenized in cultured chicken PGCs by the lipofection of the px330 plasmid and an ovomucoid gene-edited germline chimeric rooster was established [60] (Figure 2C). In the same year, transgenic chickens carrying a loxP site in the immunoglobulin heavy chain gene were successfully generated by PGCs using the CRISPR/Cas9-mediated homology-directed repair system (HDR) [61]. HDR is a cellular repair mechanism performed by the homologous recombination pathway that modifies the target genome sequence when an exogenous donor sequence is present at the sgRNA-targeted site in the CRISPR/Cas9 system. A germline chimera carrying a human interferon-β (hIFN-β) gene recombined in the ovalbumin locus was then generated by the CRISPR/Cas9-mediated HDR method, and high hIFN-β protein levels were produced in the egg white [62]. Even in basic research fields, a recent study successfully generated a genome-edited chicken targeting doublesex and mab-3-related transcription factor 1 (DMRT1) as the testis-determining gene [63].
References
- Song, G.; Han, J.Y. Avian biomodels for use as pharmaceutical bioreactors and for studying human diseases. Ann. N. Y. Acad. Sci. 2011, 1229, 69–75.
- Takahashi, Y.; Sipp, D.; Enomoto, H. Tissue interactions in neural crest cell development and disease. Science 2013, 341, 860–863.
- Naito, M. Development of avian embryo manipulation techniques and their application to germ cell manipulation. Anim. Sci. J. 2003, 74, 157–168.
- Han, J.Y. Germ cells and transgenesis in chickens. Comp. Immunol. Microbiol. Infect. Dis. 2009, 32, 61–80.
- Hillier, L.W.; Miller, W.; Birney, E.; Warren, W.; Hardison, R.C.; Ponting, C.P.; Bork, P.; Burt, D.W.; Groenen, M.A.M.; Delany, M.E.; et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716.
- Kawahara-Miki, R.; Sano, S.; Nunome, M.; Shimmura, T.; Kuwayama, T.; Takahashi, S.; Kawashima, T.; Matsuda, Y.; Yoshimura, T.; Kono, T. Next-generation sequencing reveals genomic features in the Japanese quail. Genomics 2013, 101, 345–353.
- Dalloul, R.A.; Long, J.A.; Zimin, A.V.; Aslam, L.; Beal, K.; Blomberg, L.A.; Bouffard, P.; Burt, S.W.; Crasta, O.; Croojimans, R.P.; et al. Multiplatform next-generation sequencing of the domestic turkey (Meleagris gallopavo): Genome assembly and analysis. PLoS Biol. 2010, 8, e1000475.
- Warren, W.C.; Clayton, D.F.; Ellegren, H.; Arnold, A.P.; Hillier, L.W.; Künstner, A.; Searle, S.; White, S.; Vilella, A.J.; Fairley, S.; et al. The genome of a songbird. Nature 2010, 464, 757–762.
- Schusser, B.; Collarini, E.J.; Yi, H.; Izquierdo, S.M.; Fesler, J.; Pedersen, D.; Klasing, K.C.; Kaspers, B.; Harriman, W.D.; van de Lavoir, M.C.; et al. Immunoglobulin knockout chickens via efficient homologous recombination in primordial germ cells. Proc. Natl. Acad. Sci. USA 2013, 110, 20170–20175.
- Thomas, K.R.; Capecchi, M.R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987, 51, 503–512.
- Schusser, B.; Collarini, E.J.; Pedersen, D.; Yi, H.; Ching, K.; Izquierdo, S.; Thoma, T.; Lettmann, S.; Kaspers, B.; Etches, R.J.; et al. Expression of heavy chain-only antibodies can support B-cell development in light chain knockout chickens. Eur. J. Immunol. 2016, 46, 2137–2148.
- Park, T.S.; Kang, K.S.; Han, J.Y. Current genomic editing approaches in avian transgenesis. Gen. Comp. Endocrinol. 2013, 190, 144–148.
- Park, J.S.; Lee, K.Y.; Han, J.Y. Precise genome editing in poultry and its application to industries. Genes 2020, 11, 1182.
- Lee, J.; Kim, D.H.; Lee, K. Current approaches and applications in avian genome editing. Int. J. Mol. Sci. 2020, 21, 3937.
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405.
- Garneau, J.E.; Dupuis, M.È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71.
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477.
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823.
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826.
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171.
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.H.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838.
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424.
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A.T to G.C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471.
- Bakhtiar, A.; Chowdhury, E.H. PH-responsive strontium nanoparticles for targeted gene therapy against mammary carcinoma cells. Asian J. Pharm. Sci. 2021, 16, 236–252.
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015, 33, 1293–1298.
- Cheng, H.; Zhang, F.; Ding, Y. CRISPR/Cas9 Delivery System Engineering for Genome Editing in Therapeutic Applications. Pharmaceutics 2021, 13, 1649.
- Branden, L.J.; Mohamed, A.J.; Smith, C.I.E. A peptide nucleic acid-nuclear localization signal fusion that mediates nuclear transport of DNA. Nat. Biotechnol. 1999, 17, 784–787.
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Yee, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442.
- Suzuki, Y.; Onuma, H.; Sato, R.; Sato, Y.; Hashiba, A.; Maeki, M.; Tokeshi, M.; Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K.; et al. Lipid nanoparticles loaded with ribonucleoprotein-oligonucleotide complexes synthesized using a microfluidic device exhibit robust genome editing and hepatitis B virus inhibition. J. Control. Release 2021, 330, 61–71.
- Salter, D.W.; Smith, E.J.; Hughes, S.H.; Wright, S.E.; Crittenden, L.B. Transgenic chickens: Insertion of retroviral genes into the chicken germ line. Virology 1987, 157, 236–240.
- Mizuarai, S.; Ono, K.; Yamaguchi, K.; Nishijima, K.; Kamihira, M.; Iijima, S. Production of transgenic quails with high frequency of germ-line transmission using VSV-G pseudotyped retroviral vector. Biochem. Biophys. Res. Commun. 2001, 286, 456–463.
- McGrew, M.J.; Sherman, A.; Ellard, F.M.; Lillico, S.G.; Gilhooley, H.J.; Kingsman, A.J.; Mitrophanous, K.A.; Sang, H. Efficient production of germline transgenic chickens using lentiviral vectors. EMBO Rep. 2004, 5, 728–733.
- Kamihira, M.; Ono, K.; Esaka, K.; Nishijima, K.; Kigaku, R.; Komatsu, H.; Yamashita, T.; Kyogoku, K.; Iijima, S. High-level expression of single-chain Fv-Fc fusion protein in serum and egg white of genetically manipulated chickens by using a retroviral vector. J. Virol. 2005, 79, 10864–10874.
- Chapman, S.C.; Lawson, A.; Macarthur, W.C.; Wiese, R.J.; Loechel, R.H.; Burgos-Trinidad, M.; Wakefield, J.K.; Ramabhadran, R.; Mauch, T.J.; Schoenwolf, G.C. Ubiquitous GFP expression in transgenic chicken using a lentiviral vector. Development 2005, 132, 935–940.
- Koo, B.C.; Kwon, M.S.; Choi, B.R.; Kim, J.H.; Cho, S.K.; Sohn, S.H.; Cho, E.J.; Lee, H.T.; Chang, W.; Jeon, I.; et al. Production of germline transgenic chickens expressing enhanced green fluorescent protein using a MoMLV-based retrovirus vector. FASEB J. 2006, 20, 2251–2260.
- Scott, B.B.; Lois, C. Generation of tissue-specific transgenic birds with lentiviral vectors. Proc. Natl. Acad. Sci. USA 2005, 102, 16443–16447.
- Lillico, S.G.; Sherman, A.; McGrew, M.J.; Robertson, C.D.; Smith, J.; Haslam, C.; Barnard, P.; Radcliffe, P.A.; Mitrophanous, K.A.; Elliot, E.A.; et al. Oviduct-specific expression of two therapeutic proteins in transgenic hens. Proc. Natl. Acad. Sci. USA 2007, 104, 1771–1776.
- Lee, J.; Ma, J.; Lee, K. Direct delivery of adenoviral CRISPR/Cas9 vector into the blastoderm for generation of targeted gene knockout in quail. Proc. Natl. Acad. Sci. USA 2019, 116, 13288–13292.
- Lee, J.; Kim, D.H.; Lee, K. Muscle Hyperplasia in Japanese Quail by Single Amino Acid Deletion in MSTN Propeptide. Int. J. Mol. Sci. 2020, 21, 1504.
- Xu, K.; Han, C.X.; Zhou, H.; Ding, J.M.; Xu, Z.; Yang, L.Y.; He, C.; Akinyemi, F.; Zheng, Y.M.; Qin, C.; et al. Effective MSTN Gene Knockout by AdV-Delivered CRISPR/Cas9 in Postnatal Chick Leg Muscle. Int. J. Mol. Sci. 2020, 21, 2584.
- Lee, J.; Kim, D.-H.; Karolak, M.C.; Shin, S.; Lee, K. Generation of genome-edited chicken and duck lines by adenovirus-mediated in vivo genome editing. Proc. Natl. Acad. Sci. USA 2022, 119, e2214344119.
- Pain, B.; Clark, M.E.; Shen, M.; Nakazawa, H.; Sakurai, M.; Samarut, J.; Etches, R.J. Long-term in vitro culture and characterisation of avian embryonic stem cells with multiple morphogenetic potentialities. Development 1996, 122, 2339–2348.
- Park, T.S.; Hong, Y.H.; Kwon, S.C.; Lim, J.M.; Han, J.Y. Birth of germline chimeras by transfer of chicken embryonic germ (EG) cells into recipient embryos. Mol. Reprod. Dev. 2003, 65, 389–395.
- Kim, J.N.; Park, T.S.; Park, S.H.; Park, K.J.; Kim, T.M.; Lee, S.K.; Lim, J.M.; Han, J.Y. Migration and proliferation of intact and genetically modified primordial germ cells and the generation of a transgenic chicken. Biol. Reprod. 2010, 82, 257–262.
- Swift, C.H. Origin and early history of the primordial germ cells in the chick. Am. J. Anat. 1914, 15, 483–516.
- Tsunekawa, N.; Naito, M.; Sakai, Y.; Nishida, T.; Noce, T. Isolation of chicken vasa homolog gene and tracing the origin of primordial germ cells. Development 2000, 127, 2741–2750.
- Lee, H.C.; Choi, H.J.; Lee, H.G.; Lim, J.M.; Ono, T.; Han, J.Y. DAZL expression explains origin and central formation of primordial germ cells in chickens. Stem Cells Dev. 2016, 25, 68–79.
- Murai, H.; Shibuya, M.; Kishita, R.; Sunase, C.; Tamura, K.; Saito, D. Envelopment by endothelial cells initiates translocation of avian primordial germ cell into vascular tissue. Dev. Dyn. 2021, 250, 1410–1419.
- van de Lavoir, M.C.; Diamond, J.H.; Leighton, P.A.; Mather-Love, C.; Heyer, B.S.; Bradshaw, R.; Kerchner, A.; Hooi, L.T.; Gessaro, T.M.; Swanberg, S.E.; et al. Germline transmission of genetically modified primordial germ cells. Nature 2006, 441, 766–769.
- Wentworth, B.C.; Tsai, H.; Hallett, J.H.; Gonzales, D.S.; Rajcic-Spasojevic, G. Manipulation of avian primordial germ cells and gonadal differentiation. Poult. Sci. 1989, 68, 999–1010.
- Tajima, A.; Naito, M.; Yasuda, Y.; Kuwana, T. Production of germ line chimera by transfer of primordial germ cells in the domestic chicken (Gallus domesticus). Theriogenology 1993, 40, 509–519.
- Chang, I.K.; Jeong, D.K.; Hong, Y.H.; Park, T.S.; Moon, Y.K.; Ohno, T.; Han, J.Y. Production of germline chimeric chickens by transfer of cultured primordial germ cells. Cell Biol. Int. 1997, 21, 495–499.
- Park, T.S.; Jeong, D.K.; Kim, J.N.; Song, G.; Hong, Y.H.; Lim, J.M.; Han, J.Y. Improved germline transmission in chicken chimeras produced by transplantation of gonadal primordial germ cells into recipient embryos. Biol. Reprod. 2003, 68, 1657–1662.
- Macdonald, J.; Taylor, L.; Sherman, A.; Kawakami, K.; Takahashi, Y.; Sang, H.M.; McGrew, M.J. Efficient genetic modification and germ-line transmission of primordial germ cells using piggyBac and Tol2 transposons. Proc. Natl. Acad. Sci. USA 2012, 109, 1466–1472.
- Park, T.S.; Han, J.Y. A piggyBac transposition into primordial germ cells is an efficient tool for transgenesis in chickens. Proc. Natl. Acad. Sci. USA 2012, 109, 9337–9341.
- Tyack, S.G.; Jenkins, K.A.; O’Neil, T.E.; Wise, T.G.; Morris, K.R.; Bruce, M.P.; McLeod, S.; Wade, A.J.; McKay, J.; Moore, R.J.; et al. A new method for producing transgenic birds via direct in vivo transfection of primordial germ cells. Transgenic Res. 2013, 22, 1257–1264.
- Zhu, L.; van de Lavoir, M.C.; Albanese, J.; Beenhouwer, D.O.; Cardarelli, P.M.; Cuison, S.; Deng, D.F.; Deshpande, S.; Diamond, J.H.; Green, L.; et al. Production of human monoclonal antibody in eggs of chimeric chickens. Nat. Biotechnol. 2005, 23, 1159–1169.
- Oishi, I.; Yoshii, K.; Miyahara, D.; Kagami, H.; Tagami, T. Targeted mutagenesis in chicken using CRISPR/Cas9 system. Sci. Rep. 2016, 6, 23980.
- Dimitrov, L.; Pedersen, D.; Ching, K.H.; Yi, H.; Collarini, E.J.; Izquierdo, S.; van de Lavoir, M.C.; Leighton, P.A. Germline Gene Editing in Chickens by Efficient CRISPR-Mediated Homologous Recombination in Primordial Germ Cells. PLoS ONE 2016, 11, e0154303.
- Oishi, I.; Yoshii, K.; Miyahara, D.; Tagami, T. Efficient production of human interferon β in the white of eggs from ovalbumin gene-targeted hens. Sci. Rep. 2018, 8, 10203.
- Lee, H.J.; Seo, M.; Choi, H.J.; Rengaraj, D.; Jung, K.M.; Park, J.S.; Lee, K.Y.; Kim, Y.M.; Park, K.J.; Han, S.T.; et al. DMRT1 gene disruption alone induces incomplete gonad feminization in chicken. FASEB J. 2021, 35, e21876.
More
Information
Subjects:
Developmental Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
19 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No