Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Shusei Mizushima and Version 2 by Rita Xu.
Poultry are one of the most valuable resources for human society. They are also recognized as a powerful experimental animal for basic research on embryogenesis. Demands for the supply of low-allergen eggs and bioreactors have increased with the development of programmable genome editing technology. The CRISPR/Cas9 system has recently been used to produce transgenic animals and various animals in the agricultural industry and has also been successfully adopted for the modification of chicken and quail genomes.
- CRISPR/Cas9
- intracytoplasmic sperm injection
- primordial germ cells
1. Introduction
Poultry, such as the chicken (Gallus domesticus), turkey (Meleagris gallopavo), and Japanese quail (Coturnix japonica), are commercially important species because they serve as major food sources worldwide. The poultry industry is indispensable for supporting sustainable development worldwide and is expected to significantly contribute to the United Nations’ Sustainable Development Goals. Studies on the embryos of these birds have contributed to a more detailed understanding of organogenesis [1][2][1,2] and human diseases [1], due to their oviparity. Chicken and quail embryos are easy to access and, thus, are amenable to introducing transgenes, normal or transgenic primordial germ cells (PGCs), and viruses [3][4][3,4]. The draft genome sequences of chicken, quail, turkey, and zebra finch genomes have been generated, with the information obtained supporting the generation of transgenic birds and the mass production of recombinant proteins [5][6][7][8][5,6,7,8]. The generation of gene knockout animals is a powerful approach for identifying essential proteins and the functions of uncharacterized genes in many species, as is the case in birds. The combination of recent technical advances is expected to provide novel insights into future aspects of avian biotechnology as well as human society.
The generation of gene knockout chickens by homologous recombination using PGC-mediated methods was initially reported in 2013 [9] following its establishment in mice [10]. Immunoglobulin light chain knockout chickens were subsequently generated using the same method [11]. Although these achievements contributed to the progression of avian gene targeting, the efficiency of this recombination was very low. In this regard, the emergence of site-specific nuclease has provided new avenues for modifications to avian genomes [12][13][14][12,13,14]. The earliest programmable genome editing tools for the generation of gene knockout animals were zinc finger nuclease (ZFN) and transcription activator-like effector nuclease (TALEN) [15]. These enzymes were artificially created by the fusion of FoKI endonucleases with the capacity to recognize long-chain DNA. FoKI endonucleases recognize target DNA and induce double-strand breaks (DSB) and small indels by error-prone non-homologous end joining (NHEJ).
2. CRISPR/Cas9-Mediated Genome Editing Technology
The CRISPR/Cas system, originally found in bacteria and archaea, is an RNA-based adaptive immune system that destroys invading plasmids, phages, and viruses [16][17][18][19,20,21]. The nucleoprotein complex, consisting of three crucial components: CRISPR-coding RNA (crRNA), trans-activating crRNA (tracrRNA), and Cas protein, recognize exogenous DNA and degrade it by endonuclease activity [19][22], causing error-prone NHEJ. The binding site of Cas9 is located upstream of the protospacer adjacent motif (PAM) containing the 5′-NGG base sequence. Humanized Cas9 protein or Cas9 derived from Streptococcus pyogenes Cas9 (SpCas9), combined with a synthetic single guide RNA (sgRNA) produced by fusing crRNA with tracrRNA, has been shown to trigger DSB in mammalian cells [20][21][23,24]. This CRISPR/Cas9 system may be easily designed and prepared for a plasmid vector by changing the sgRNA sequence to a specific region of the genome sequence instead of ZFN and TALEN described above, and its use has rapidly expanded. The recent re-engineering of Cas9 has resulted in the establishment of dead Cas9 (dCas9) with mutations in two nuclease domains, which has extended the application of the CRISPR/Cas9 system with the potential transcriptional inhibitor/activator, and point mutations [22][23][24][25][25,26,27,28] (Figure 1A).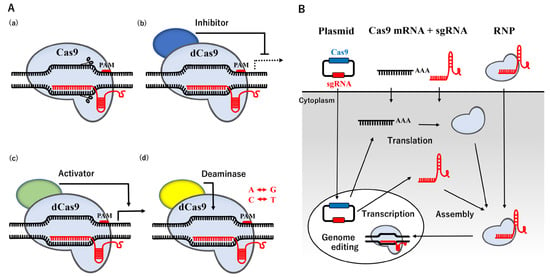
Figure 1. Applications and Routes of the CRISPR-Cas9 system. (A) Schematic illustration of the applications. (a) A basic molecular model of the CRISPR/Cas9 system. (b) dCas9 fused with a transcriptional inhibitor (blue) represses transcription. (c) dCas9 fused with a transcriptional activator (green) boosts transcription. (d) dCas9 bound to adenine or cytosine deaminase (yellow) modifies A to G or T to C, respectively. (B) Schematic illustration of the routes. Three forms of plasmids encoding Cas9 and sgRNA, the RNAs of Cas9 mRNA and sgRNA, or a complex of Cas9 protein and sgRNA are available for delivery into cells by transfection or a microinjection.
3. Current Approaches for Avian Genome Editing Based on the CRISPR/Cas9 System
Avian genome editing technology is based on a procedure for establishing transgenic chicken lines. Various approaches have been attempted to create transgenic birds, including viral infection, the chimeric method, and sperm-mediated gene transfer (Figure 2).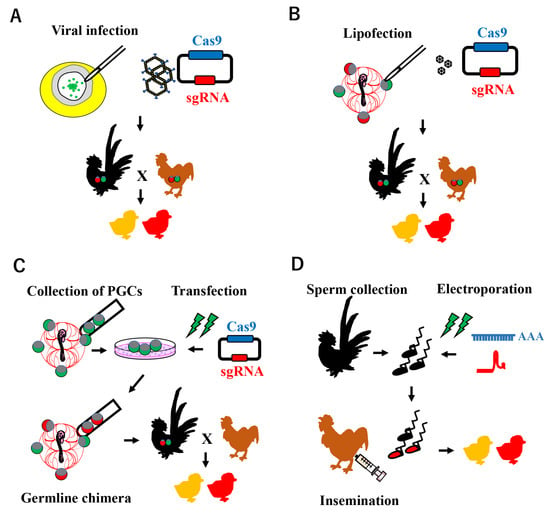
Figure 2. Schematic illustration of CRISPR-Cas9 system-mediated genome editing in poultry. (A) Viral infection model: a recombinant adenovirus carrying CRISPR/Cas9 components is infected at blastoderm stage X, where PGCs exist in the center area. (B) PGC-mediated genome editing: the plasmid-encoding Cas9 and sgRNA expression cassettes with lipofectamine are microinjected into embryonic blood vessels, in which the PGCs are circulating. (C) PGC-mediated genome editing: the CRISPR/Cas9 system is introduced into cultured PGCs in vitro, which are mainly collected from embryonic blood vessels. Enriched genome-edited PGCs are transferred into the blood vessels of recipient embryos to generate a germline chimera. (D) Sperm Transfection-Assisted Gene Editing (STAGE): a mixture of Cas9 mRNA and sgRNA is transfected into ejaculated sperm collected from roosters, which are then subjected to artificial insemination in hens.