Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohsen Yoosefzadeh Najafabadi | -- | 4202 | 2023-06-13 17:39:04 | | | |
| 2 | Lindsay Dong | Meta information modification | 4202 | 2023-06-15 03:33:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yoosefzadeh Najafabadi, M.; Hesami, M.; Rajcan, I. Non-Mendelian Heredity in Plant Breeding. Encyclopedia. Available online: https://encyclopedia.pub/entry/45522 (accessed on 22 July 2026).
Yoosefzadeh Najafabadi M, Hesami M, Rajcan I. Non-Mendelian Heredity in Plant Breeding. Encyclopedia. Available at: https://encyclopedia.pub/entry/45522. Accessed July 22, 2026.
Yoosefzadeh Najafabadi, Mohsen, Mohsen Hesami, Istvan Rajcan. "Non-Mendelian Heredity in Plant Breeding" Encyclopedia, https://encyclopedia.pub/entry/45522 (accessed July 22, 2026).
Yoosefzadeh Najafabadi, M., Hesami, M., & Rajcan, I. (2023, June 13). Non-Mendelian Heredity in Plant Breeding. In Encyclopedia. https://encyclopedia.pub/entry/45522
Yoosefzadeh Najafabadi, Mohsen, et al. "Non-Mendelian Heredity in Plant Breeding." Encyclopedia. Web. 13 June, 2023.
Copy Citation
Mendelian heredity is the cornerstone of plant breeding and has been used to develop new varieties of plants since the 19th century. However, there are several breeding cases, such as cytoplasmic inheritance, methylation, epigenetics, hybrid vigor, and loss of heterozygosity (LOH), where Mendelian heredity is not applicable, known as non-Mendelian heredity. This type of inheritance can be influenced by several factors besides the genetic architecture of the plant and its breeding potential. Therefore, exploring various non-Mendelian heredity mechanisms, their prevalence in plants, and the implications for plant breeding is of paramount importance to accelerate the pace of crop improvement.
Beavis effect
chromosomal rearrangements
cytoplasmic inheritance
epigenetics
hybridization
loss of heterozygosity
polyploidy
1. Introduction
The field of plant breeding and genetics has traditionally been based on the work of Gregor Mendel, who first proposed the ‘laws of inheritance’ [1]. The Mendelian laws of inheritance, also known as Mendelian genetics, state that the inheritance of traits in an offspring is determined by the combination of discrete units of heredity, called genes, which are passed on from parent to offspring [2]. Mendelian genetics is based on the idea that each gene is passed on in a certain predictable way and can be used to explain the inheritance of different traits, which can be supported by three laws of inheritance proposed by Mendel: the Law of Segregation, Law of Dominance, and Law of Independent Assortment [2][3].
The Law of Dominance states that when two versions of a gene (alleles) are present, one allele will be expressed, while the other allele will be “masked” or not expressed [4][5], which is known as the dominant and recessive alleles, respectively. The Law of Segregation states that during the formation of gametes, the two copies of each gene separate so that each gamete only contains one copy of the gene [6][7]. This means that the offspring will receive only one copy of the gene from each parent, thus leading to the phenotype of the offspring being determined by the combination of the alleles from both parents [7]. The Law of Independent Assortment states that the alleles at different loci segregate independently of each other, meaning that the inheritance of one allele at a locus does not influence the inheritance of the alleles at other loci [6][7].
The Mendelian laws of inheritance can provide sufficient explanations for Monogenic inheritance (a single gene determining a single trait) traits such as seed shape, flower color, and seed coat color [7][8]. However, there are multiple examples that Mendelian laws of inheritance are not applicable for polygenic inheritance (multiple genes at different loci are involved in determining a single trait) traits such as yield, maturity, and abiotic/biotic stresses [9][10]. Those traits that are not explained by Mendel’s rules, are referred to as non-Mendelian caused by non-Mendelian heredity [8][10]. Non-Mendelian traits may be transmitted from one generation to the next in a number of different ways, such as through the action of gene-environment and/or gene–gene interactions, or even through epigenetic changes [8][9].
Gene-environment interaction is one of the most important factors in plant breeding areas when the expression of a gene is influenced by the environment [11][12]. In plant breeding, environmental factors can play a role in the expression of complex traits such as yield [13]. In plant breeding, non-Mendelian traits can be transmitted through gene–gene interactions, either through epistatic (complex) interactions between genes, or through the action of regulators of gene expression [8][9][11].
Non-Mendelian traits can be used in plant breeding to create novel varieties with desired characteristics. The identification of these traits and the ability to generate novel varieties through the manipulation of these traits is a major focus of most research in plant breeding [14][15][16].
2. The Basis of Non-Mendelian Heredity
Non-Mendelian heredity is a term used to describe inheritance patterns that do not fit the classical Mendelian inheritance model, which allows breeders to create new varieties of plants with desired characteristics in a different way [2][3][6]. By understanding the various forms of non-Mendelian heredity, breeders can better control the genetics of the plants they are working with and create varieties that are more likely to be successful [6][10]. The lack of clear-cut Mendelian inheritance patterns in plants is largely because many plants are capable of producing offspring through different reproductive strategies such as cross-pollination, self-pollination, and asexual reproduction [6][8][9][10]. This complexity results in multiple patterns of inheritance that do not fit the traditional Mendelian model.
Cross-pollination can result in offspring with characteristics that are a combination of the parental forms. This is known as hybridization and can be used to create new varieties of plants with desired traits [17][18]. However, the offspring may not always be identical to the parental forms, as the genetic material from both parents can mix and recombine (genetic recombination) in unpredictable ways [19]. Understanding the implications of non-Mendelian heredity is also important, as it can lead to a lack of genetic diversity and the appearance of harmful recessive traits [19]. Self-pollination is when a plant produces pollen and fertilizes itself, mostly resulting in offspring genetically identical to the parent [20]. While self-pollination can be used to maintain desirable traits in a species, it can also lead to inbreeding, which can reduce genetic diversity and lead to the appearance of recessive traits [21]. Asexual reproduction is another form of non-Mendelian heredity [22]. This is when a single parent reproduces by cloning itself, resulting in offspring that are genetically identical to the parent. Asexual reproduction is often used to propagate desirable traits in plants, as it allows for a greater degree of control over the genetic makeup of the offspring [22][23].
Using genetic information generated by different methods such as pedigree analysis, DNA sequence analysis, linkage analysis, and genetic mapping are some of the common ways to calculate the genetic recombination rate in plants [24]. Genetic information is arranged in chromosomes of various lengths, resulting in the genetic linkage of genes [25]. For example, in green pea (Pisum sativum) as the main crop Mendel worked on, there are seven chromosomes, of which two and three of the genes encoding Mendel’s traits are located on chromosomes 1 or 4, respectively [26]. It is remarkable that most trait combinations indicated unlinked genes, which is explained by the large size of the pea chromosomes and the distance of the loci, leading to a high recombination frequency and no linkage disequilibrium [27][28]. If the genes were more closely linked, it would have made it difficult to observe and interpret new combinations as the result of independent segregation [28]. Mendel was able to accurately characterize the material he had available and avoided the issue of polygenic traits determined by multiple genes (quantitative inheritance) [26].
3. Polyploidy
Non-Mendelian heredity can also be observed in plants through polyploidy [18]. This is when a species has more than two sets of chromosomes, resulting in offspring that is genetically distinct from their parents [18]. Autopolyploidy and allopolyploidy are two of the most common ploidy in plants [29]. Autopolyploidy occurs when an individual has multiple sets of chromosomes from the same species, whereas allopolyploidy is when the individual has multiple sets of chromosomes from different species [29][30]. Some researchers distinguish between two types of polyploidy based on their origins (parentage), while others focus on genetic characteristics such as chromosomal profile and behavior [31]. Polysomic polyploids are formed when duplicated chromosomes are completely homologous and result from multivalent or random bivalent segregation during meiosis [32]. Disomic polyploids occur when duplicated chromosomes are partially homologous and strictly from bivalent homologous chromosomes [33]. Despite differences in origin, both types of polyploids have high levels of gene duplication and heterozygosity, with autopolyploids having higher levels of heterozygosity than diploids due to outcrossing [32][33]. This phenomenon can be used to create new varieties of plants with desired traits such as increased size, higher yields, or higher disease resistance, but it can also lead to reduced fertility, as the offspring may not be able to produce viable gametes [32].
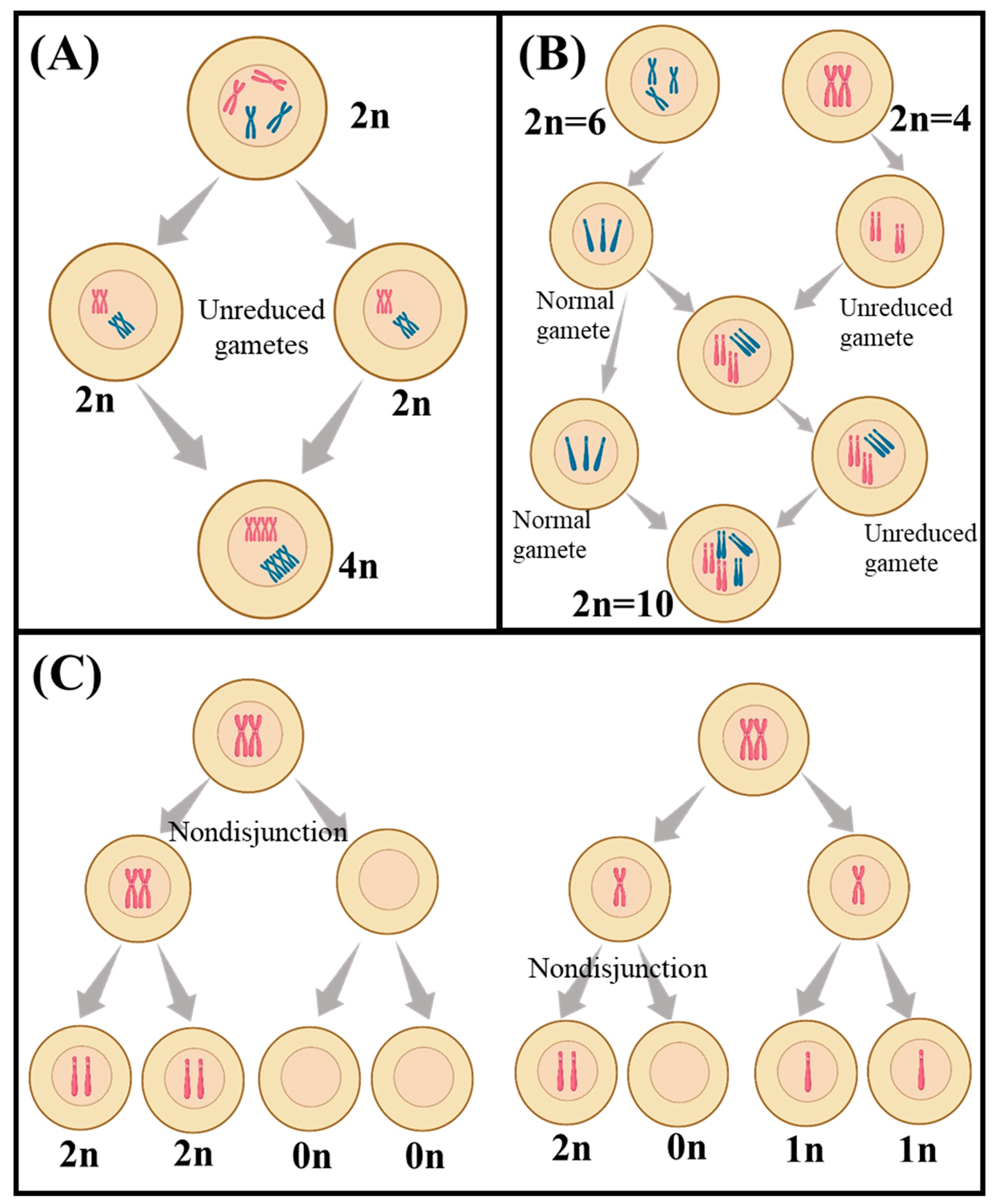
3.1. Autopolyploidy and Allopolyploidy
Meiosis in polyploids is more complex than in diploids, but the expected segregation ratios can still be calculated. In autotetraploid species (Figure 1A), such as the potato, which has four sets of chromosomes from the same species, a recessive trait would be expressed in only one out of every 36 F2 plants [34]. This frequency could be further reduced in autohexaploid plants, with six copies of each chromosome, to one in 64 [34]. Additionally, autopolyploid plants have the potential for double reduction, which is when two recombined chromosomes move to the same pole in anaphase I, a process not seen in diploids [35]. The fact that recessive traits are expressed at different frequencies in autopolyploid and allopolyploid species (Figure 2B) is important for breeders and researchers, which allows them to predict how often are certain traits expressed in a given generation, which can help inform breeding strategies and help identify desirable traits [36][37].
Figure 1. A schematic view of different polyploidization including (A) autopolyploidy, (B) allopolyploidy, and (C) aneuploidy. The schematic was created by using BioRender.com.
3.2. Aneupolyploidy
Aneuploidy refers to the condition where the number of chromosomes in a cell is not an exact multiple of the haploid number of the species [38] (Figure 1C). Aneuploidy can occur naturally in plants, but it can also be induced by breeding or through the use of chemical or radiation treatments [39]. Aneuploids are often characterized by reduced fertility and can be used to generate new plants with desirable traits, as well as to study the effects of chromosome changes on plant development and physiology [38][39].
3.3. Heterosomes and B-Chromosomes
Heterosomes, which are sex chromosomes in dioecious plants, can also be responsible for the expression of different traits, in addition to the type of flower (sex) [40]. One example is the Silene latifolia, a species of flowering plant in the Caryophyllaceae family, which exhibits differences in its flower morphology based on the heterosomes it expresses [40]. The female plants have larger petals and sepals and more white flowers than the male plants, which have smaller petals and sepals and fewer white flowers [40].
4. Cytoplasmic Inheritance
4.1. Cytoplasmic Male Sterility (CMS)
CMS is considered as a type of non-Mendelian inheritance because it is inherited solely through the cytoplasm of the female parent and is not determined by the genes of either parent, in which the male reproductive organs of the plant are non-functional [41][42]. It is a form of genetic male sterility and is used extensively in plant breeding programs to produce hybrid varieties [42][43]. The trait is caused by a mutation in the mitochondrial genome which results in the production of aberrant proteins that interfere with the normal function of the male reproductive organs. CMS is used to produce hybrid varieties of crop plants by crossing a sterile CMS variety with a fertile restorer variety [43][44]. The hybrid progenies are all fertile, and this hybrid vigor results in increased yields and improved disease resistance.
4.2. Mitochondrial Inheritance
In recent years, mitochondrial inheritance has been used increasingly in plant breeding to create improved varieties of crops with higher yields, improved disease resistance, and improved nutritional value [45]. By introducing specific gene mutations into the plant’s mitochondria, breeders can control the expression and activity of certain genes and alter the plant’s phenotype. Plant mitochondrial genomes are larger and less conserved than chloroplast genomes, therefore, have received less attention [46].
The use of mitochondrial mutations in plant breeding has been gaining attention in recent years as a tool for improving a variety of plant traits. Rauf [47] reported the use of mitochondrial mutations to create a drought-tolerant sunflower, resulting in an increase in the amount of unsaturated fatty acids in the plant’s seed oil, which increased the sunflower’s tolerance to drought stress. Similarly, mitochondrial mutations were used to increase the yield of rice plants by increasing the amount of photosynthetic efficiency [48]. These mutations also increased the plant’s tolerance to heat stress [48]. Mitochondrial mutations were successfully applied to change the flower color of petunia with a mutation in the mitochondrial gene’s coding region [49].
4.3. Chloroplast Inheritance
Chloroplast inheritance in plant breeding refers to the transmission of chloroplast DNA from the female parent to all of the progeny of a cross [50]. Chloroplasts are organelles within the cells of plants that contain their own genetic material, known as plastomes. These plastomes can be inherited from a female parent, allowing breeders to track the maternal parent in a cross, and can be used to develop male-sterile lines [50][51]. Chloroplast genomes in plants are highly conserved sequences of 100–150 Kb containing around 100 genes [52]. The standard structure of a chloroplast is composed of four elements, including inverted repeats that divide the large and small single-copy regions [53]. They have been a popular choice for plant identification due to their high copy numbers in the cell [54]. Previous methods of chloroplast isolation or PCR amplification were challenged by the same sequences existing in both the nuclear and mitochondrial genomes [55][56][57].
5. Chromosomal Rearrangements
Chromosomal rearrangements are a type of genetic alteration that can be used in plant breeding programs to produce new varieties with desirable traits [58]. Rearrangements involve breaking and rejoining sections of chromosomes, resulting in rearrangements of gene order, deletions or additions of genetic material, or changes in chromosome structure [58][59]. These changes can lead to new combinations of genes that can confer desirable traits. Chromosomal rearrangements were successfully implemented in different breeding programs to increase the genetic variation of wheat [60], maize [61], and rice [62]. Zhang et al. [62] examined the effects of multiple DNA double-strand breaks (DSBs) in rice plants and found that rice varieties with a high number of simultaneous DSBs (e.g., over 50) showed low-frequency large chromosomal deletions and duplications, but this was not the case for plants with lower order DSBs (e.g., under 10). Therefore, large chromosomal rearrangement can occur in varieties with a large number of DSBs [62].
6. Gene–Gene Interaction
Gregor Mendel conducted dihybrid crosses to examine how genes can affect traits. In Gregor Mendel’s experiments, he crossed a homozygous plant with round and yellow seeds (RRYY) with another homozygous plant with wrinkled and green seeds (rryy) and observed a phenotypic ratio of 9:3:3:1, where each gene locus had an independent effect on a single phenotype [6]. Nevertheless, in numerous instances, complex phenotypes do not adhere to the principles of segregation and independent assortment elucidated by Mendelian genetics, as they are frequently governed by the contribution of multiple genes to their ultimate expression [63]. When two genes contribute to the same phenotype (gene–gene interaction), the phenotypic ratio may deviate from that expected from the independent action of each gene, a phenomenon known as epistasis [64]. Such interactions between two or more loci can create novel phenotypes for which the allelic effects of single genes are described as “dominant” and “recessive” [64]. Epistasis is a phenotypic-level phenomenon, wherein an independent assortment of genotypes is observed, yet the phenotypic outcomes may differ from the anticipated ratios [64].
Shull [65] seminal study of the weedy plant Bursa bursa-pastoris, more commonly known as Shepard’s Purse, is a classic example of epistasis. Upon crossbreeding doubly heterozygous plants, Shull observed a ratio of 15:1 between triangular and oval capsules, respectively [65]. This phenomenon is thought to be the result of two pathways, each containing a dominant locus that produces the triangular shape [65]. When both pathways are blocked by recessive alleles, an oval-shaped seed capsule is produced, a phenomenon known as recessive-by-recessive interaction [65]. This suggests that having two recessive genotypes results in a different phenotype than having just one from either locus.
Epistatic interactions between quantitative traits can manifest in two forms: a change in the magnitude of the effects or a change in the direction of the effects [64]. In the absence of epistasis, the estimates of the additive and dominance effects at each locus remain the same regardless of the genotype of the other locus [64][66]. However, with epistasis, the effect of one locus depends on the genotype at its interacting locus [66]. There is still much debate about the relevance of epistasis to quantitative traits, with some concentrating on individual genotypes and others focusing on the epistatic genetic variance in populations [66]. Genetical epistasis is independent of allele frequencies, whereas the total genetic variance in a population is divided between additive, dominance, and epistatic variance, which are all based on allele frequencies [67]. Epistasis can cause different effects in populations because the effect of one locus is dependent on the allele frequency of another locus. Its influence can be strong in one population and weak or even reversed in another [67].
Most genetic variance that is observed for quantitative traits is additive, which could be either ‘real’ or ‘apparent’ due to epistatic gene action at many loci [68]. This is significant for the purposes of heritability and predicting phenotypes; however, it is especially important when trying to understand the effects of genetic drift and inbreeding, as well as the genotype-phenotype map, long-term responses to selection, and genetic interactions [68].
Epistasis can be studied through the examination of mutants in the same homozygous genetic background [69]. Epistasis occurs if the difference in phenotype between the double mutant cannot be predicted by the combined effects of the two single mutants [70]. This can either be negative or synergistic, meaning that the double mutant is more mutant than expected, or positive, meaning that the double mutant is less mutant than expected [69][70].
In QTL mapping, epistasis can be estimated by a statistical model with factors for each QTL and the interaction between them [64]. Multifactorial perturbations can be used to screen for epistasis with a small number of individuals, which is more efficient than constructing all possible gene combinations [64]. Power to detect epistasis is highest in inbred lines due to the equal frequencies of each allele [71]. However, in small mapping populations, the number of individuals with rare homozygous genotypes is small, which increases the variance of the phenotype [64]. Additionally, other loci can produce confounding effects, and multiple testing can make it difficult to detect epistasis. Most studies only assess additive effects, but epistatic effects can be as large as main effects and can occur between non-significant loci [71][72]. Epistatic interactions have been observed in genetic studies of growth rate and metabolites in A. thaliana [73] and differences in inflorescence and whole-plant architecture in maize and teosinte [74]. These findings demonstrate that epistasis must be considered in order to understand the genetics of complex traits.
By introgressing fragments of DNA from one genotype into the genetic background of another, it is possible to create a powerful QTL mapping design [75]. This can be done either by introducing entire chromosomes or with smaller fragments across the genome. While only a small number of introgression lines are necessary, they can be used to map QTLs with high accuracy [75][76]. Epistasis occurs when the combined effect of the introgressed fragments is not the same as the average difference in phenotype between the two parental strains [64][76]. Epistatic interactions between loci can lead to distinct main effects of each locus, as well as a failure to replicate estimated QTL effects when allele frequencies between populations vary [75].
7. Epigenetics
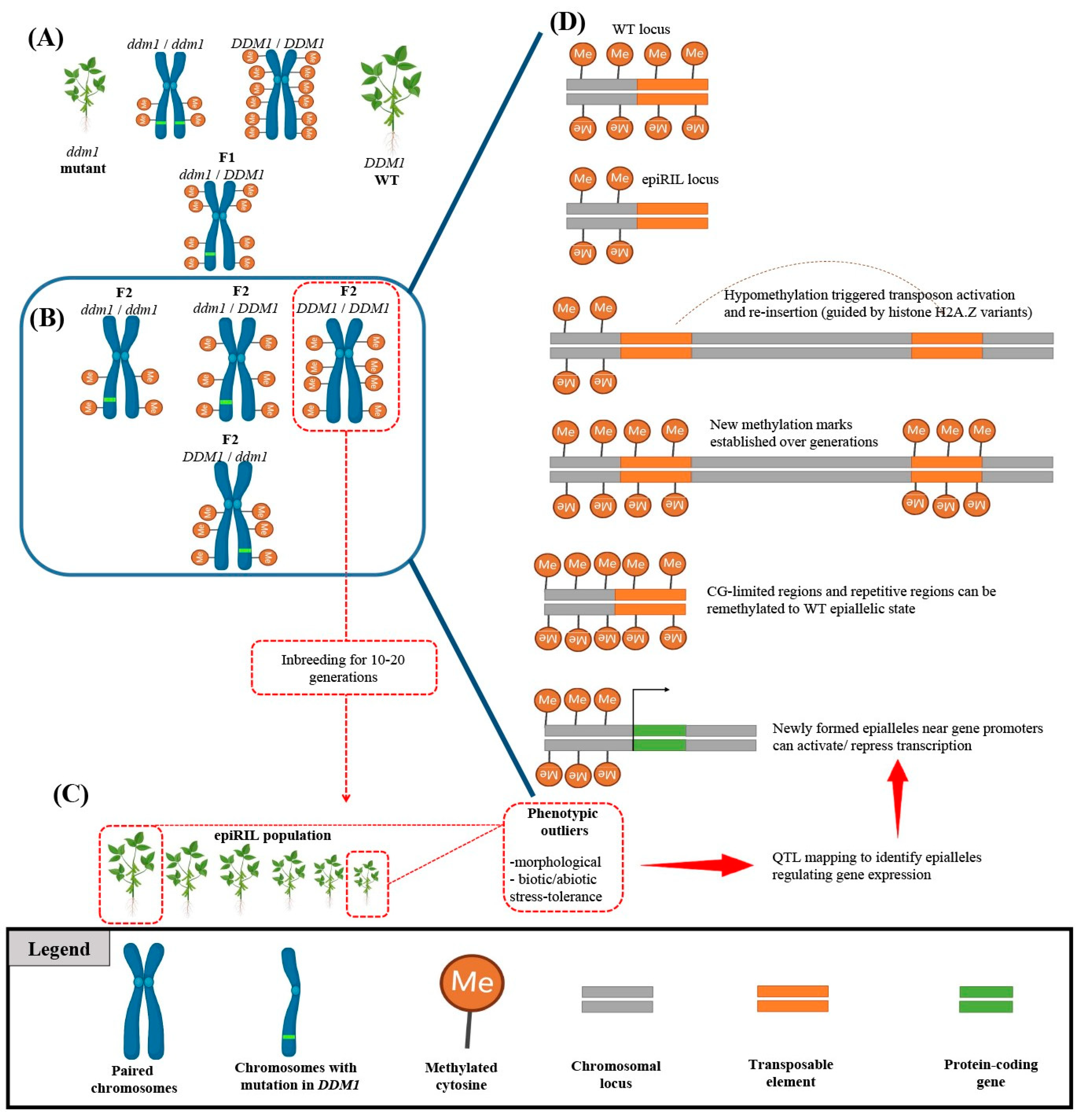
Epigenetic modifications (e.g., DNA methylation, chromatin remodeling, histone modification, and RNA-directed DNA methylation) can be defined as any changes in gene expression without alterations in the DNA sequence [77]. It is well-documented that epigenetics plays a fundamental role in plant growth and development by regulating gene expression [77][78][79][80][81]. Furthermore, the discovery of epialleles, which can be defined as genetic variations caused by changes in DNA methylation, has opened up a new understanding of how epigenetic modifications can lead to novel phenotypes and contribute to evolution [82]. Since epigenetic modifications can be influenced by a range of environmental stressors, the epigenetic state of an individual can be highly plastic and can be influenced by both internal and external factors, leading to epialleles being transmitted to offspring without following traditional Mendelian inheritance patterns [83][84][85]. This non-Mendelian behavior of epialleles has important implications for understanding and studying inheritance, as well as for the fields of evolution and ecology [85][86][87]. It also has practical implications for plant breeding, where understanding the non-Mendelian inheritance of epialleles can help to develop crops that are better adapted to changing environmental conditions and exhibit improved yield potential through epigenetic recombinant inbred lines (epiRILs) (Figure 2) [87][88].
Figure 2. Identification of novel epialleles using epigenetic recombinant inbred line (epiRIL) generation. (A) Crossing wild-type (WT) plants with DNA methylation-deficient mutants such as ddm1 mutant can redistribute genome-wide methylation patterns. (B) Progeny carrying WT alleles are selected for multigenerational inbreeding to generate epigenetic recombinant inbred lines (epiRILs). (C) The epiRIL population is evaluated for variations in stress resistance or morphological traits to identify phenotypic outliers. (D) The identified epiRIL lines undergo an epigenetic quantitative trait loci (epiQTL) analysis to discover novel epialleles. Such epialleles can occur due to the activation of transposable elements (TEs) and their reinsertion into distant loci, determined by chromatin properties and the nature of the target sequence (CG content). The scheme was adapted from Srikant and Tri Wibowo [89] and was created by using BioRender.com.
8. Gene-Environment Interaction
In most breeding programs, the inheriting genetic traits are not determined by the laws of Mendelian genetics, because of the interaction of environmental factors and the genetic makeup of the plant. Gene-environment interaction (GEI) refers to the effect of the environment on the expression of genetic variation, and it is known to play an important role in the heritability of traits in plants. Most of the complex traits that are significantly under control with environmental factors have lower heritability than others that are less affected by the environment and mostly affected by genetics.
Lower heritability traits in plants can have several disadvantages, which can have a negative impact on the pace of breeding program. One of the main disadvantages of lower heritability traits in plants is that they are more difficult to select for [90]. When selecting for traits, it is generally easier to select for high heritability traits, as they are more likely to be passed on to the next generation (i.e., follow Mendelian genetics). Low heritability traits, on the other hand, are more likely to be changed during the selection process, as they are less likely to be passed on to the next generation.
9. Linkage and Association Mapping in Plant Breeding
Soon after discovering Mendelian heredity, several breeders demonstrated that some traits in their crosses did not adhere to Mendel’s principles of heredity and seemed “coupled” [91]. To explain this phenomenon, scientists proposed a hypothesis that certain traits must be inherited together, e.g., through the linkage of certain genes [91][92]. This hypothesis was later verified through further experiments, which determined that certain alleles were always inherited together [91]. This phenomenon was then referred to as genetic linkage. In genetics, linkage refers to the tendency of certain genes or genetic markers to appear together more often than expected by chance [91]. Linkage occurs when two or more genes are located close to each other on the same chromosome.
If genetic linkage is prevalent in the plant genomes, why did Mendel not detect it through his experiments on pea plants? Mendel studied seven genes in pea plants, which have seven chromosomes. Although Mendel did not select gene pairs that always resided on separate chromosomes, some of the gene pairs studied by Mendel were found to be located on the same chromosome [2].
Crossing genetically different parents is the initial step in generating linkage maps and locating genes related to the desired trait. Different types of genetic populations have been formed for mapping traits, such as F2, F2:3, backcross introgression lines (BILs), recombinant inbred lines (RILs), near-isogenic lines (NILs), multiparent advanced generation intercross (MAGIC) populations, and association mapping populations based on natural populations [93]. Bulk segregant analysis (BSA), F2, and backcross populations are commonly used in short-term molecular mapping populations, but RILs, NILs, doubled haploid (DH), nested association mapping (NAM), and MAGIC populations, can be used for more precise phenotyping and sharing between breeders over a longer period of time [94].
10. Loss of Heterozygosity (LOH)
Loss of heterozygosity (LOH) is an important phenomenon in plant breeding, as it can lead to significant changes in the genetic composition of a population [95]. LOH is a type of non-Mendelian heredity that occurs when a plant loses one of its two alleles due to the mutation of a gene [95][96]. When this happens, the plant will no longer have two copies of a particular gene and will only have a single copy of that gene [96]. This, in turn, can lead to a decrease in the genetic diversity of the population, as well as a reduction in the effectiveness of selection. In plant breeding, the formation of homozygous lines through inbreeding can lead to eliminating some alleles, resulting in reduced genetic diversity. Furthermore, the outcrossing of related varieties can also lead to LOH, as the offspring will not inherit a full complement of alleles from both parents.
Loss-of-heterozygosity was reported in somatic cells of rice hybrids for the first time by Wang et al. [97], which involves the selected plant ‘AMR’, of the Chinese rice cultivar ‘ZhongxinNo.1′, as one parent. Variations were identified in the vegetative parts of the same plant using random amplified polymorphic DNA (RAPD) markers and molecular assays [97]. All F2 panicle rows from F1 hybrids involving AMR became fixed for all assayed RAPD markers, and this genotype fixation was confirmed by field observations of the F3 progenies [97]. The results suggested that in these hybrids, both parental homologues of some chromosomes in somatic cells are not always present. Later, Wang et al. [98] proposed a new biological mechanism called ‘assortment mitosis’, to explain this phenomenon. This mechanism can develop uniform progenies as early as the F2 generation and shorten the time required to obtain fixed non-parental type progenies for subsequent performance trials [98].
References
- Priyadarshan, P. Plant Breeding: Classical to Modern; Springer: Singapore, 2019.
- Allen, G.E. Mendel and modern genetics: The legacy for today. Endeavour 2003, 27, 63–68.
- Gautam, A. MendelTs Laws. In Encyclopedia of Animal Cognition and Behavior; Springer: Cham, Switzerland, 2018.
- Marks, J. The construction of Mendel’s laws. Evol. Anthropol. Issues News Rev. 2008, 17, 250–253.
- Zhang, J. What Has Genomics Taught An Evolutionary Biologist? Genom. Proteom. Bioinform. 2023; In Press.
- Patwardhan, D. Mendelian Principle of Inheritance. In Genetics Fundamentals Notes; Springer Nature Singapore: Singapore, 2022; pp. 53–83.
- Mackay, T.F.; Anholt, R.R. Gregor Mendel’s legacy in quantitative genetics. PLoS Biol. 2022, 20, e3001692.
- Xu, S. Review of Mendelian Genetics. In Quantitative Genetics; Springer: Cham, Switzerland, 2022; pp. 13–24.
- Wolf, J.B.; Ferguson-Smith, A.C.; Lorenz, A. Mendel’s laws of heredity on his 200th birthday: What have we learned by considering exceptions? Heredity 2022, 129, 1–3.
- Jessop, A. Mendel in and after His Time. 2022. Available online: http://philsci-archive.pitt.edu/20332/ (accessed on 9 May 2023).
- Chung, K.P.; Gonzalez-Duran, E.; Ruf, S.; Endries, P.; Bock, R. Control of plastid inheritance by environmental and genetic factors. Nat. Plants 2023, 9, 68–80.
- Yoosefzadeh-Najafabadi, M.; Rajcan, I.; Vazin, M. High-throughput plant breeding approaches: Moving along with plant-based food demands for pet food industries. Front. Vet. Sci. 2022, 9, 991844.
- Yoosefzadeh Najafabadi, M.; Hesami, M.; Eskandari, M. Machine Learning-Assisted Approaches in Modernized Plant Breeding Programs. Genes 2023, 14, 777.
- Chen, C.; Wang, M.; Zhu, J.; Tang, Y.; Zhang, H.; Zhao, Q.; Jing, M.; Chen, Y.; Xu, X.; Jiang, J. Long-term effect of epigenetic modification in plant–microbe interactions: Modification of DNA methylation induced by plant growth-promoting bacteria mediates promotion process. Microbiome 2022, 10, 36.
- Graudal, L.; Dawson, I.K.; Hale, I.; Powell, W.; Hendre, P.; Jamnadass, R. ‘Systems approach’plant breeding illustrated by trees. Trends Plant Sci. 2022, 27, 158–165.
- Bowerman, A.F.; Byrt, C.S.; Roy, S.J.; Whitney, S.M.; Mortimer, J.C.; Ankeny, R.A.; Gilliham, M.; Zhang, D.; Millar, A.A.; Rebetzke, G.J. Potential abiotic stress targets for modern genetic manipulation. Plant Cell 2023, 35, 139–161.
- Burson, B.L.; Young, B.A. Breeding and improvement of tropical grasses. In Tropical Forage Plants: Development and Use; CRC Press: Boca Raton, FL, USA, 2000; pp. 59–80.
- Ranney, T.G. Polyploidy: From Evolution to New Plant Development. pp. 137–142. Available online: https://ena.ipps.org/uploads/docs/56_85.pdf (accessed on 9 May 2023).
- Linder, C.R.; Rieseberg, L.H. Reconstructing patterns of reticulate evolution in plants. Am. J. Bot. 2004, 91, 1700–1708.
- Schoen, D.J.; Lloyd, D.G. Self-and cross-fertilization in plants. III. Methods for studying modes and functional aspects of self-fertilization. Int. J. Plant Sci. 1992, 153, 381–393.
- Bradshaw, J.E. Breeding Diploid F1 Hybrid Potatoes for Propagation from Botanical Seed (TPS): Comparisons with Theory and Other Crops. Plants 2022, 11, 1121.
- De Meeûs, T.; Prugnolle, F.; Agnew, P. Asexual reproduction: Genetics and evolutionary aspects. Cell. Mol. Life Sci. 2007, 64, 1355–1372.
- Cornaro, L.; Banfi, C.; Cucinotta, M.; Colombo, L.; van Dijk, P.J. Asexual Reproduction through Seeds: The Complex Case of Diplosporous Apomixis; Oxford University Press: Oxford, UK, 2023.
- Yoosefzadeh-Najafabadi, M.; Rajcan, I.; Eskandari, M. Optimizing genomic selection in soybean: An important improvement in agricultural genomics. Heliyon 2022, 8, e11873.
- van Rengs, W.M.; Schmidt, M.H.W.; Effgen, S.; Le, D.B.; Wang, Y.; Zaidan, M.W.A.M.; Huettel, B.; Schouten, H.J.; Usadel, B.; Underwood, C.J. A chromosome scale tomato genome built from complementary PacBio and Nanopore sequences alone reveals extensive linkage drag during breeding. Plant J. 2022, 110, 572–588.
- Ellis, T.N.; Hofer, J.M.; Timmerman-Vaughan, G.M.; Coyne, C.J.; Hellens, R.P. Mendel, 150 years on. Trends Plant Sci. 2011, 16, 590–596.
- Blixt, S. Why didn’t Gregor Mendel find linkage? Nature 1975, 256, 206.
- Mittelsten Scheid, O. Mendelian and non-Mendelian genetics in model plants. Plant Cell 2022, 34, 2455–2461.
- Barker, M.S.; Arrigo, N.; Baniaga, A.E.; Li, Z.; Levin, D.A. On the relative abundance of autopolyploids and allopolyploids. New Phytol. 2016, 210, 391–398.
- Soltis, D.E.; Soltis, P.S.; Schemske, D.W.; Hancock, J.F.; Thompson, J.N.; Husband, B.C.; Judd, W.S. Autopolyploidy in angiosperms: Have we grossly underestimated the number of species? Taxon 2007, 56, 13–30.
- Soltis, D.E.; Buggs, R.J.; Doyle, J.J.; Soltis, P.S. What we still don’t know about polyploidy. Taxon 2010, 59, 1387–1403.
- Scott, A.D.; Van de Velde, J.D.; Novikova, P.Y. Inference of polyploid origin and inheritance mode from population genomic data. In Polyploidy: Methods and Protocols; Springer: New York, NY, USA, 2023; pp. 279–295.
- Osborn, T.C.; Pires, J.C.; Birchler, J.A.; Auger, D.L.; Chen, Z.J.; Lee, H.-S.; Comai, L.; Madlung, A.; Doerge, R.; Colot, V. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 2003, 19, 141–147.
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846.
- Levings III, C.; Alexander, D. Double reduction in autotetraploid maize. Genetics 1966, 54, 1297.
- Te Beest, M.; Le Roux, J.J.; Richardson, D.M.; Brysting, A.K.; Suda, J.; Kubešová, M.; Pyšek, P. The more the better? The role of polyploidy in facilitating plant invasions. Ann. Bot. 2012, 109, 19–45.
- Gallais, A. Quantitative Genetics and Breeding Methods in Autopolyploid Plants; Inra: Versailles, France, 2003; pp. 1–516.
- Orr, B.; Godek, K.M.; Compton, D. Aneuploidy. Curr. Biol. 2015, 25, R538–R542.
- Zhu, J.; Tsai, H.-J.; Gordon, M.R.; Li, R. Cellular stress associated with aneuploidy. Dev. Cell 2018, 44, 420–431.
- Hobza, R.; Hudzieczek, V.; Kubat, Z.; Cegan, R.; Vyskot, B.; Kejnovsky, E.; Janousek, B. Sex and the flower–developmental aspects of sex chromosome evolution. Ann. Bot. 2018, 122, 1085–1101.
- Vinod, K. Cytoplasmic genetic male sterility in plants. A molecular perspective. In Proceedings of the Training Programme on Advances and Accomplishments in Heteron Breeding; Tamil Nadu Agricultural University: Coimbotore, India, 2005.
- Toriyama, K. Molecular basis of cytoplasmic male sterility and fertility restoration in rice. Plant Biotechnol. 2021, 38, 285–295.
- Melonek, J.; Duarte, J.; Martin, J.; Beuf, L.; Murigneux, A.; Varenne, P.; Comadran, J.; Specel, S.; Levadoux, S.; Bernath-Levin, K. The genetic basis of cytoplasmic male sterility and fertility restoration in wheat. Nat. Commun. 2021, 12, 1036.
- Xu, F.; Yang, X.; Zhao, N.; Hu, Z.; Mackenzie, S.A.; Zhang, M.; Yang, J. Exploiting sterility and fertility variation in cytoplasmic male sterile vegetable crops. Hortic. Res. 2022, 9, uhab039.
- Morales, F.; Ancín, M.; Fakhet, D.; González-Torralba, J.; Gámez, A.L.; Seminario, A.; Soba, D.; Ben Mariem, S.; Garriga, M.; Aranjuelo, I. Photosynthetic metabolism under stressful growth conditions as a bases for crop breeding and yield improvement. Plants 2020, 9, 88.
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120.
- Rauf, S. Breeding strategies for sunflower (Helianthus annuus L.) genetic improvement. Adv. Plant Breed. Strateg. Ind. Food Crops 2019, 6, 637–673.
- Luo, D.; Xu, H.; Liu, Z.; Guo, J.; Li, H.; Chen, L.; Fang, C.; Zhang, Q.; Bai, M.; Yao, N. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577.
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16, S154–S169.
- Park, H.-S.; Lee, W.K.; Lee, S.-C.; Lee, H.O.; Joh, H.J.; Park, J.Y.; Kim, S.; Song, K.; Yang, T.-J. Inheritance of chloroplast and mitochondrial genomes in cucumber revealed by four reciprocal F1 hybrid combinations. Sci. Rep. 2021, 11, 2506.
- Heinke, L. Chilling paternal chloroplasts. Nat. Rev. Mol. Cell Biol. 2023, 24, 166.
- Dobrogojski, J.; Adamiec, M.; Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant. 2020, 42, 98.
- Henry, R.J.; Rice, N.; Edwards, M.; Nock, C.J. Next-generation technologies to determine plastid genome sequences. Chloroplast Biotechnol. Methods Protoc. 2014, 1132, 39–46.
- Nock, C.J.; Waters, D.L.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011, 9, 328–333.
- Ananda, G.; Norton, S.; Blomstedt, C.; Furtado, A.; Møller, B.; Gleadow, R.; Henry, R. Phylogenetic relationships in the Sorghum genus based on sequencing of the chloroplast and nuclear genes. Plant Genome 2021, 14, e20123.
- Brozynska, M.; Copetti, D.; Furtado, A.; Wing, R.A.; Crayn, D.; Fox, G.; Ishikawa, R.; Henry, R.J. Sequencing of Australian wild rice genomes reveals ancestral relationships with domesticated rice. Plant Biotechnol. J. 2017, 15, 765–774.
- Healey, A.; Lee, D.J.; Furtado, A.; Henry, R.J. Evidence of inter-sectional chloroplast capture in Corymbia among sections Torellianae and Maculatae. Aust. J. Bot. 2018, 66, 369–378.
- Rönspies, M.; Schindele, P.; Puchta, H. CRISPR/Cas-mediated chromosome engineering: Opening up a new avenue for plant breeding. J. Exp. Bot. 2021, 72, 177–183.
- Prieto, P. Chromosome manipulation for plant breeding purposes. Agronomy 2020, 10, 1695.
- Badaeva, E.; Dedkova, O.; Gay, G.; Pukhalskyi, V.; Zelenin, A.; Bernard, S.; Bernard, M. Chromosomal rearrangements in wheat: Their types and distribution. Genome 2007, 50, 907–926.
- Sharma, S.P.; Peterson, T. Complex chromosomal rearrangements induced by transposons in maize. Genetics 2023, 223, iyac124.
- Zhang, Y.; Wu, Y.; Li, G.; Qi, A.; Zhang, Y.; Zhang, T.; Qi, Y. Genome-wide investigation of multiplexed CRISPR-Cas12a-mediated editing in rice. Plant Genome 2022, e20266.
- Halfhill, M.D.; Warwick, S.I. Mendelian genetics and plant reproduction. In Plant Biotechnology and Genetics: Principles, Techniques and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; Volume 34.
- Mackay, T.F. Epistasis and quantitative traits: Using model organisms to study gene–gene interactions. Nat. Rev. Genet. 2014, 15, 22–33.
- Shull, G.H. Duplicate genes for capsule-form in Bursa bursa-pastoris. Z Indukt Abstamm Vererb. 1914, 12, 97–149.
- Carlborg, Ö.; Haley, C.S. Epistasis: Too often neglected in complex trait studies? Nat. Rev. Genet. 2004, 5, 618–625.
- Phillips, P.C. Epistasis—The essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 2008, 9, 855–867.
- Hayes, B.J.; Lewin, H.A.; Goddard, M.E. The future of livestock breeding: Genomic selection for efficiency, reduced emissions intensity, and adaptation. Trends Genet. 2013, 29, 206–214.
- Wang, R.; Lammers, M.; Tikunov, Y.; Bovy, A.G.; Angenent, G.C.; de Maagd, R.A. The rin, nor and Cnr spontaneous mutations inhibit tomato fruit ripening in additive and epistatic manners. Plant Sci. 2020, 294, 110436.
- Evans, K.S.; van Wijk, M.H.; McGrath, P.T.; Andersen, E.C.; Sterken, M.G. From QTL to gene: C. elegans facilitates discoveries of the genetic mechanisms underlying natural variation. Trends Genet. 2021, 37, 933–947.
- Deutschbauer, A.M.; Davis, R.W. Quantitative trait loci mapped to single-nucleotide resolution in yeast. Nat. Genet. 2005, 37, 1333–1340.
- Gerke, J.; Lorenz, K.; Cohen, B. Genetic interactions between transcription factors cause natural variation in yeast. Science 2009, 323, 498–501.
- Rowe, H.C.; Hansen, B.G.; Halkier, B.A.; Kliebenstein, D.J. Biochemical networks and epistasis shape the Arabidopsis thaliana metabolome. Plant Cell 2008, 20, 1199–1216.
- Doebley, J.; Stec, A.; Gustus, C. teosinte branched1 and the origin of maize: Evidence for epistasis and the evolution of dominance. Genetics 1995, 141, 333–346.
- Mackay, T.F. Epistasis for quantitative traits in Drosophila. Epistasis Methods Protoc. 2015, 1253, 47–70.
- Shao, H.; Burrage, L.C.; Sinasac, D.S.; Hill, A.E.; Ernest, S.R.; O’Brien, W.; Courtland, H.-W.; Jepsen, K.J.; Kirby, A.; Kulbokas, E. Genetic architecture of complex traits: Large phenotypic effects and pervasive epistasis. Proc. Natl. Acad. Sci. USA 2008, 105, 19910–19914.
- Gallusci, P.; Agius, D.R.; Moschou, P.N.; Dobránszki, J.; Kaiserli, E.; Martinelli, F. Deep inside the epigenetic memories of stressed plants. Trends Plant Sci. 2023, 28, 142–153.
- Ramakrishnan, M.; Papolu, P.K.; Satish, L.; Vinod, K.K.; Wei, Q.; Sharma, A.; Emamverdian, A.; Zou, L.-H.; Zhou, M. Redox status of the plant cell determines epigenetic modifications under abiotic stress conditions and during developmental processes. J. Adv. Res. 2022, 42, 99–116.
- Sobral, M.; Sampedro, L. Phenotypic, epigenetic, and fitness diversity within plant genotypes. Trends Plant Sci. 2022, 27, 843–846.
- Hesami, M.; Jones, A.M.P. Potential roles of epigenetic memory on the quality of clonal cannabis plants: Content and profile of secondary metabolites. Med. Usage Cannabis Cannabinoids 2023, 1, 1–14.
- Lloyd, J.P.B.; Lister, R. Epigenome plasticity in plants. Nat. Rev. Genet. 2022, 23, 55–68.
- Kalisz, S.; Purugganan, M.D. Epialleles via DNA methylation: Consequences for plant evolution. Trends Ecol. Evol. 2004, 19, 309–314.
- Zhang, Y.; Wendte, J.M.; Ji, L.; Schmitz, R.J. Natural variation in DNA methylation homeostasis and the emergence of epialleles. Proc. Natl. Acad. Sci. USA 2020, 117, 4874–4884.
- Kakutani, T. Epi-Alleles in Plants: Inheritance of Epigenetic Information over Generations. Plant Cell Physiol. 2002, 43, 1106–1111.
- Weigel, D.; Colot, V. Epialleles in plant evolution. Genome Biol. 2012, 13, 249.
- House, M.; Lukens, L. The Role of Germinally Inherited Epialleles in Plant Breeding: An Update. In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications: Transcriptional Regulation and Chromatin Remodelling in Plants; Alvarez-Venegas, R., De-la-Peña, C., Casas-Mollano, J.A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 115–128. Available online: https://link.springer.com/chapter/10.1007/978-3-030-14760-0_3 (accessed on 6 March 2023).
- Hudzieczek, V.; Hobza, R.; Cápal, P.; Šafář, J.; Doležel, J. If Mendel Was Using CRISPR: Genome Editing Meets Non-Mendelian Inheritance. Adv. Funct. Mater. 2022, 32, 2202585.
- Casas, E.; Vavouri, T. Mechanisms of epigenetic inheritance of variable traits through the germline. Reproduction 2020, 159, R251–R263.
- Srikant, T.; Tri Wibowo, A. The Underlying Nature of Epigenetic Variation: Origin, Establishment, and Regulatory Function of Plant Epialleles. Int. J. Mol. Sci. 2021, 22, 8618.
- Yoosefzadeh Najafabadi, M.; Rajcan, I. Six Decades of Soybean Breeding in Ontario, Canada: A Tradition of Innovation. Can. J. Plant Sci. 2022.
- Stoltenberg, S.F. Foundations of Behavior Genetics; Cambridge University Press: Cambridge, UK, 2022.
- Darden, L. Theory construction in genetics. In Scientific Discovery: Case Studies; Springer: Dordrecht, The Netherlands, 1980; pp. 151–170.
- Sinha, S.; Kushwaha, B.K.; Deshmukh, R.K. QTL Mapping Using Advanced Mapping Populations and High-throughput Genotyping. Genotyping by Sequencing for Crop Improvement; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2022; pp. 52–79.
- Rani, K.; Kumar, M.; Razzaq, A.; Ajay, B.; Kona, P.; Bera, S.K.; Wani, S.H. Recent advances in molecular marker technology for QTL mapping in plants. In QTL Mapping in Crop Improvement; Academic Press: Cambridge, MA, USA, 2023; pp. 1–15.
- Heil, C.S.S.; DeSevo, C.G.; Pai, D.A.; Tucker, C.M.; Hoang, M.L.; Dunham, M.J. Loss of heterozygosity drives adaptation in hybrid yeast. Mol. Biol. Evol. 2017, 34, 1596.
- Tutaj, H.; Pirog, A.; Tomala, K.; Korona, R. Genome-scale patterns in the loss of heterozygosity incidence in Saccharomyces cerevisiae. Genetics 2022, 221, iyac032.
- Wang, R.R.-C.; Li, X.-M.; Chatterton, N. Loss of heterozygosity and accelerated genotype fixation in rice hybrids. Genome 1999, 42, 789–796.
- Wang, R.R.-C.; Li, X.-M.; Chatterton, N.J. A proposed mechanism for loss of heterozygosity in rice hybrids. Euphytica 2001, 118, 119–126.
More
Information
Subjects:
Agriculture, Dairy & Animal Science
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Revisions:
2 times
(View History)
Update Date:
15 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No