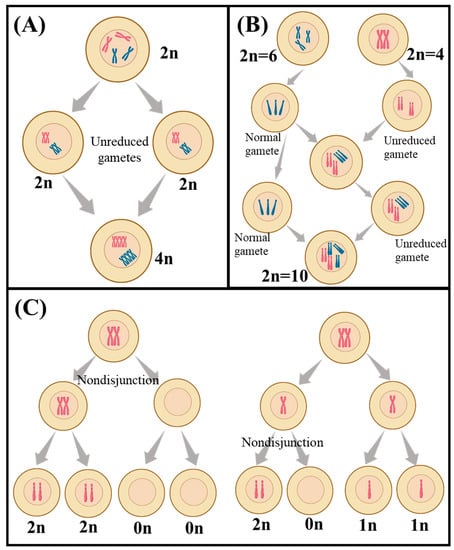
Chromosomal rearrangements are a type of genetic alteration that can be used in plant breeding programs to produce new varieties with desirable traits [
71]. Rearrangements involve breaking and rejoining sections of chromosomes, resulting in rearrangements of gene order, deletions or additions of genetic material, or changes in chromosome structure [
71,
72]. These changes can lead to new combinations of genes that can confer desirable traits. Chromosomal rearrangements were successfully implemented in different breeding programs to increase the genetic variation of wheat [
73], maize [
74], and rice [
75]. Zhang et al. [
75] examined the effects of multiple DNA double-strand breaks (DSBs) in rice plants and found that rice varieties with a high number of simultaneous DSBs (e.g., over 50) showed low-frequency large chromosomal deletions and duplications, but this was not the case for plants with lower order DSBs (e.g., under 10). Therefore, large chromosomal rearrangement can occur in varieties with a large number of DSBs [
75].
6. Gene–Gene Interaction
Gregor Mendel conducted dihybrid crosses to examine how genes can affect traits. In Gregor Mendel’s experiments, he crossed a homozygous plant with round and yellow seeds (RRYY) with another homozygous plant with wrinkled and green seeds (rryy) and observed a phenotypic ratio of 9:3:3:1, where each gene locus had an independent effect on a single phenotype [
6]. Nevertheless, in numerous instances, complex phenotypes do not adhere to the principles of segregation and independent assortment elucidated by Mendelian genetics, as they are frequently governed by the contribution of multiple genes to their ultimate expression [
77]. When two genes contribute to the same phenotype (gene–gene interaction), the phenotypic ratio may deviate from that expected from the independent action of each gene, a phenomenon known as epistasis [
78]. Such interactions between two or more loci can create novel phenotypes for which the allelic effects of single genes are described as “dominant” and “recessive” [
78]. Epistasis is a phenotypic-level phenomenon, wherein an independent assortment of genotypes is observed, yet the phenotypic outcomes may differ from the anticipated ratios [
78].
Shull [
79] seminal study of the weedy plant Bursa bursa-pastoris, more commonly known as Shepard’s Purse, is a classic example of epistasis. Upon crossbreeding doubly heterozygous plants, Shull observed a ratio of 15:1 between triangular and oval capsules, respectively [
79]. This phenomenon is thought to be the result of two pathways, each containing a dominant locus that produces the triangular shape [
79]. When both pathways are blocked by recessive alleles, an oval-shaped seed capsule is produced, a phenomenon known as recessive-by-recessive interaction [
79]. This suggests that having two recessive genotypes results in a different phenotype than having just one from either locus.
Epistatic interactions between quantitative traits can manifest in two forms: a change in the magnitude of the effects or a change in the direction of the effects [
78]. In the absence of epistasis, the estimates of the additive and dominance effects at each locus remain the same regardless of the genotype of the other locus [
78,
83]. However, with epistasis, the effect of one locus depends on the genotype at its interacting locus [
83]. There is still much debate about the relevance of epistasis to quantitative traits, with some concentrating on individual genotypes and others focusing on the epistatic genetic variance in populations [
83]. Genetical epistasis is independent of allele frequencies, whereas the total genetic variance in a population is divided between additive, dominance, and epistatic variance, which are all based on allele frequencies [
84]. Epistasis can cause different effects in populations because the effect of one locus is dependent on the allele frequency of another locus. Its influence can be strong in one population and weak or even reversed in another [
84].
Most genetic variance that is observed for quantitative traits is additive, which could be either ‘real’ or ‘apparent’ due to epistatic gene action at many loci [
85]. This is significant for the purposes of heritability and predicting phenotypes; however, it is especially important when trying to understand the effects of genetic drift and inbreeding, as well as the genotype-phenotype map, long-term responses to selection, and genetic interactions [
85].
Epistasis can be studied through the examination of mutants in the same homozygous genetic background [
86]. Epistasis occurs if the difference in phenotype between the double mutant cannot be predicted by the combined effects of the two single mutants [
87]. This can either be negative or synergistic, meaning that the double mutant is more mutant than expected, or positive, meaning that the double mutant is less mutant than expected [
86,
87].
In QTL mapping, epistasis can be estimated by a statistical model with factors for each QTL and the interaction between them [
78]. Multifactorial perturbations can be used to screen for epistasis with a small number of individuals, which is more efficient than constructing all possible gene combinations [
78]. Power to detect epistasis is highest in inbred lines due to the equal frequencies of each allele [
91]. However, in small mapping populations, the number of individuals with rare homozygous genotypes is small, which increases the variance of the phenotype [
78]. Additionally, other loci can produce confounding effects, and multiple testing can make it difficult to detect epistasis. Most studies only assess additive effects, but epistatic effects can be as large as main effects and can occur between non-significant loci [
91,
92]. Epistatic interactions have been observed in genetic studies of growth rate and metabolites in
A. thaliana [
93] and differences in inflorescence and whole-plant architecture in maize and teosinte [
94]. These findings demonstrate that epistasis must be considered in order to understand the genetics of complex traits.
By introgressing fragments of DNA from one genotype into the genetic background of another, it is possible to create a powerful QTL mapping design [
96]. This can be done either by introducing entire chromosomes or with smaller fragments across the genome. While only a small number of introgression lines are necessary, they can be used to map QTLs with high accuracy [
96,
97]. Epistasis occurs when the combined effect of the introgressed fragments is not the same as the average difference in phenotype between the two parental strains [
78,
97]. Epistatic interactions between loci can lead to distinct main effects of each locus, as well as a failure to replicate estimated QTL effects when allele frequencies between populations vary [
96].
7. Epigenetics
Epigenetic modifications (e.g., DNA methylation, chromatin remodeling, histone modification, and RNA-directed DNA methylation) can be defined as any changes in gene expression without alterations in the DNA sequence [
103]. It is well-documented that epigenetics plays a fundamental role in plant growth and development by regulating gene expression [
103,
104,
105,
106,
107]. Furthermore, the discovery of epialleles, which can be defined as genetic variations caused by changes in DNA methylation, has opened up a new understanding of how epigenetic modifications can lead to novel phenotypes and contribute to evolution [
108]. Since epigenetic modifications can be influenced by a range of environmental stressors, the epigenetic state of an individual can be highly plastic and can be influenced by both internal and external factors, leading to epialleles being transmitted to offspring without following traditional Mendelian inheritance patterns [
109,
110,
111]. This non-Mendelian behavior of epialleles has important implications for understanding and studying inheritance, as well as for the fields of evolution and ecology [
111,
112,
113]. It also has practical implications for plant breeding, where understanding the non-Mendelian inheritance of epialleles can help to develop crops that are better adapted to changing environmental conditions and exhibit improved yield potential through epigenetic recombinant inbred lines (epiRILs) (
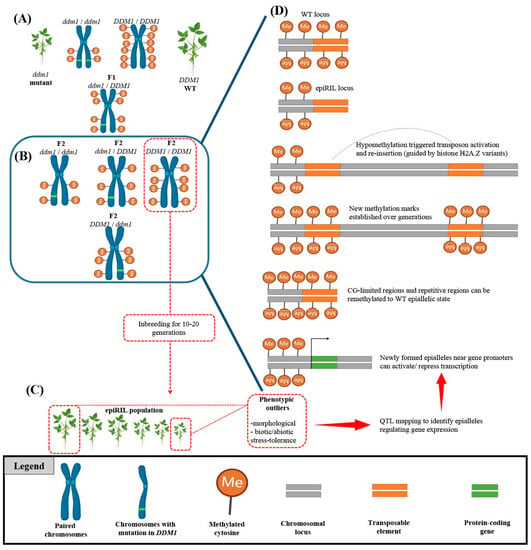
Figure 2) [
113,
114].
Figure 2. Identification of novel epialleles using epigenetic recombinant inbred line (epiRIL) generation. (
A) Crossing wild-type (WT) plants with DNA methylation-deficient mutants such as ddm1 mutant can redistribute genome-wide methylation patterns. (
B) Progeny carrying WT alleles are selected for multigenerational inbreeding to generate epigenetic recombinant inbred lines (epiRILs). (
C) The epiRIL population is evaluated for variations in stress resistance or morphological traits to identify phenotypic outliers. (
D) The identified epiRIL lines undergo an epigenetic quantitative trait loci (epiQTL) analysis to discover novel epialleles. Such epialleles can occur due to the activation of transposable elements (TEs) and their reinsertion into distant loci, determined by chromatin properties and the nature of the target sequence (CG content). The scheme was adapted from Srikant and Tri Wibowo [
115] and was created by using BioRender.com.
8. Gene-Environment Interaction
In most breeding programs, the inheriting genetic traits are not determined by the laws of Mendelian genetics, because of the interaction of environmental factors and the genetic makeup of the plant. Gene-environment interaction (GEI) refers to the effect of the environment on the expression of genetic variation, and it is known to play an important role in the heritability of traits in plants. Most of the complex traits that are significantly under control with environmental factors have lower heritability than others that are less affected by the environment and mostly affected by genetics.
Lower heritability traits in plants can have several disadvantages, which can have a negative impact on the pace of breeding program. One of the main disadvantages of lower heritability traits in plants is that they are more difficult to select for [
127]. When selecting for traits, it is generally easier to select for high heritability traits, as they are more likely to be passed on to the next generation (i.e., follow Mendelian genetics). Low heritability traits, on the other hand, are more likely to be changed during the selection process, as they are less likely to be passed on to the next generation.
9. Linkage and Association Mapping in Plant Breeding
Soon after discovering Mendelian heredity, several breeders demonstrated that some traits in their crosses did not adhere to Mendel’s principles of heredity and seemed “coupled” [
128]. To explain this phenomenon, scientists proposed a hypothesis that certain traits must be inherited together, e.g., through the linkage of certain genes [
128,
129]. This hypothesis was later verified through further experiments, which determined that certain alleles were always inherited together [
128]. This phenomenon was then referred to as genetic linkage. In genetics, linkage refers to the tendency of certain genes or genetic markers to appear together more often than expected by chance [
128]. Linkage occurs when two or more genes are located close to each other on the same chromosome.
If genetic linkage is prevalent in the plant genomes, why did Mendel not detect it through his experiments on pea plants? Mendel studied seven genes in pea plants, which have seven chromosomes. Although Mendel did not select gene pairs that always resided on separate chromosomes, some of the gene pairs studied by Mendel were found to be located on the same chromosome [
2].
Crossing genetically different parents is the initial step in generating linkage maps and locating genes related to the desired trait. Different types of genetic populations have been formed for mapping traits, such as F
2, F
2:3, backcross introgression lines (BILs), recombinant inbred lines (RILs), near-isogenic lines (NILs), multiparent advanced generation intercross (MAGIC) populations, and association mapping populations based on natural populations [
130]. Bulk segregant analysis (BSA), F
2, and backcross populations are commonly used in short-term molecular mapping populations, but RILs, NILs, doubled haploid (DH), nested association mapping (NAM), and MAGIC populations, can be used for more precise phenotyping and sharing between breeders over a longer period of time [
131].
10. Loss of Heterozygosity (LOH)
Loss of heterozygosity (LOH) is an important phenomenon in plant breeding, as it can lead to significant changes in the genetic composition of a population [
153]. LOH is a type of non-Mendelian heredity that occurs when a plant loses one of its two alleles due to the mutation of a gene [
153,
154]. When this happens, the plant will no longer have two copies of a particular gene and will only have a single copy of that gene [
154]. This, in turn, can lead to a decrease in the genetic diversity of the population, as well as a reduction in the effectiveness of selection. In plant breeding, the formation of homozygous lines through inbreeding can lead to eliminating some alleles, resulting in reduced genetic diversity. Furthermore, the outcrossing of related varieties can also lead to LOH, as the offspring will not inherit a full complement of alleles from both parents.
Loss-of-heterozygosity was reported in somatic cells of rice hybrids for the first time by Wang et al. [
155], which involves the selected plant ‘AMR’, of the Chinese rice cultivar ‘ZhongxinNo.1′, as one parent. Variations were identified in the vegetative parts of the same plant using random amplified polymorphic DNA (RAPD) markers and molecular assays [
155]. All F
2 panicle rows from F
1 hybrids involving AMR became fixed for all assayed RAPD markers, and this genotype fixation was confirmed by field observations of the F
3 progenies [
155]. The results suggested that in these hybrids, both parental homologues of some chromosomes in somatic cells are not always present. Later, Wang et al. [
156] proposed a new biological mechanism called ‘assortment mitosis’, to explain this phenomenon. This mechanism can develop uniform progenies as early as the F
2 generation and shorten the time required to obtain fixed non-parental type progenies for subsequent performance trials [
156].