 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Michael Pusch | -- | 2526 | 2023-06-13 12:11:44 | | | |
| 2 | Jessie Wu | + 3 word(s) | 2529 | 2023-06-14 05:35:34 | | |
Video Upload Options
Endosomes and lysosomes are intracellular vesicular organelles with important roles in cell functions such as protein homeostasis, clearance of extracellular material, and autophagy. Endolysosomes are characterized by an acidic luminal pH that is critical for proper function. Five members of the gene family of voltage-gated ChLoride Channels (CLC proteins) are localized to endolysosomal membranes, carrying out anion/proton exchange activity and thereby regulating pH and chloride concentration. Mutations in these vesicular CLCs cause global developmental delay, intellectual disability, various psychiatric conditions, lysosomal storage diseases, and neurodegeneration, resulting in severe pathologies or even death.
1. Introduction—The ChLoride Channel Family
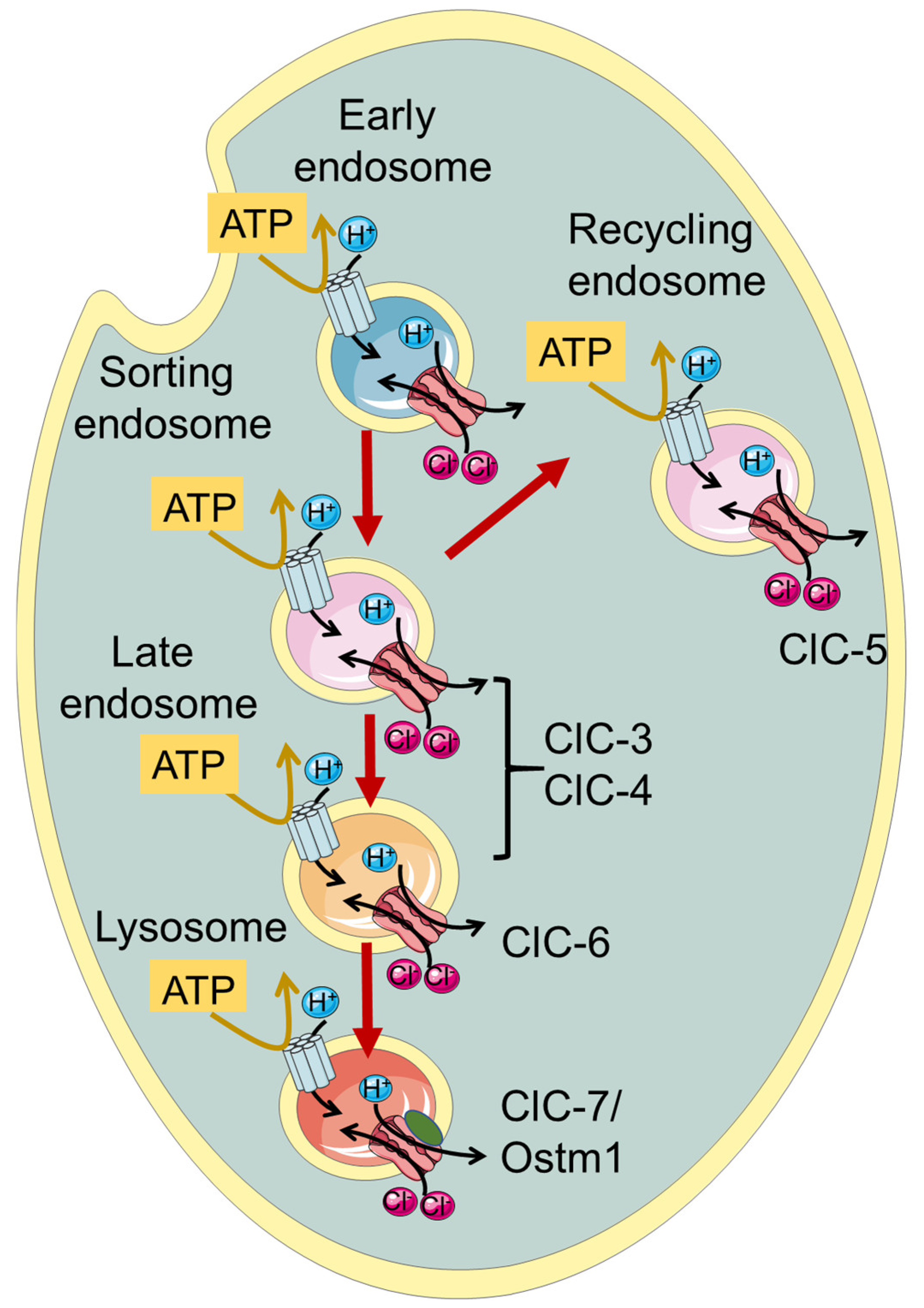
| Protein | Gene | Cellular Localization | Neurological Disorder | Symptoms | References |
|---|---|---|---|---|---|
| ClC-3 | CLCN3 | Sorting and late endosomes | Global developmental delay | Intellectual disability, agenesis of the corpus callosum, epilepsy, visual impairment, hypotonia, anxiety, dysmorphic facial features |
[39] |
| ClC-4 | CLCN4 | Sorting and late endosomes | CLCN4-related X linked intellectual disability syndrome | Intellectual disability, epilepsy, autism, growth and feeding difficulties, epilepsy, movement disorders, gastrointestinal conditions, dysmorphic facial features | [40][41][42] |
| ClC-6 | CLCN6 | Late endosomes | |||
| Early Onset Neurodegeneration | Severe neurodegeneration, severe generalized hypotonia and respiratory insufficiency, brain atrophy | [43] | |||
| Kufs’ disease | Adult-onset neuronal ceroid Lipofuscinosis, movement and cognitive function impairment |
[44][45] | |||
| West syndrome | Severe developmental delay, autism, movement disorder, microcephaly, facial dysmorphism, visual impairment |
[46] | |||
| ClC-7/ Ostm1 |
CLCN7/ OSTM1 |
Lysosomes | |||
| Autosomal Recessive Osteopetrosis | Osteopetrosis, lysosomal storage disease, neurodegeneration, visual impairment | [38][47][48] | |||
| Gain of function CLCN7 related disease | Delayed myelination and development, organomegaly, and hypopigmentation | [49] |
2. ClC-3 and ClC-4
While ClC-4 KO mice have no overt phenotype, in 2013 and 2016, patients (mostly pediatric) with a range of neurodevelopmental and psychiatric complications have been described with X-linked CLCN4 variants [40,42,65] (see Table 1). In heterologous expression, these and some novel variants [66] showed variable loss of function effects. It is important to note that complete loss of ClC-4 protein leads to non-syndromic intellectual disability in males and no disease in heterozygous females. In contrast, de novo and inherited missense variants can lead to severe syndromic neurological disease in males as well as in females, suggesting a dominant effect. In a more recent study, a large number of CLCN4 families was investigated, describing a large spectrum of clinical phenotypes and studying > 50 missense variants in heterologous expression [67]. Novel biophysical mechanisms were discovered for new and already described variants. These included a toxic gain of function characterized by the presence of negative currents at acidic extracellular (luminal) pH, and a shift in the voltage dependence of gating to more positive voltages [67]. Both effects can be expected to exert dominant negative effects in ClC-3/ClC-4 heterodimers.
Almost simultaneously came the discovery of the first variants in CLCN3 that cause global developmental delay, intellectual disability and neurodevelopmental disorders [39] (Table 1). Detailed functional analysis revealed a toxic gain of function for two missense variants, similar to the above-described effects in some CLCN4 variants [39].
3. ClC-6
4. ClC-7
References
- Jentsch, T.J.; Pusch, M. CLC chloride channels and transporters: Structure, function, physiology, and disease. Physiol. Rev. 2018, 98, 1493–1590.
- Jentsch, T.J.; Steinmeyer, K.; Schwarz, G. Primary structure of Torpedo marmorata chloride channel isolated by expression cloning in Xenopus oocytes. Nature 1990, 348, 510–514.
- Accardi, A.; Miller, C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature 2004, 427, 803–807.
- Leisle, L.; Ludwig, C.F.; Wagner, F.A.; Jentsch, T.J.; Stauber, T. ClC-7 is a slowly voltage-gated 2Cl−/1H+-exchanger and requires Ostm1 for transport activity. EMBO J. 2011, 30, 2140–2152.
- Middleton, R.E.; Pheasant, D.J.; Miller, C. Homodimeric architecture of a ClC-type chloride ion channel. Nature 1996, 383, 337–340.
- Ludewig, U.; Pusch, M.; Jentsch, T.J. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature 1996, 383, 340–343.
- Maduke, M.; Pheasant, D.J.; Miller, C. High-level expression, functional reconstitution, and quaternary structure of a prokaryotic ClC-type chloride channel. J. Gen. Physiol. 1999, 114, 713–722.
- Mindell, J.A.; Maduke, M.; Miller, C.; Grigorieff, N. Projection structure of a ClC-type chloride channel at 6.5 Å resolution. Nature 2001, 409, 219–223.
- Dutzler, R.; Campbell, E.B.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature 2002, 415, 287–294.
- Dutzler, R.; Campbell, E.B.; MacKinnon, R. Gating the selectivity filter in ClC chloride channels. Science 2003, 300, 108–112.
- Ponting, C.P. CBS domains in CIC chloride channels implicated in myotonia and nephrolithiasis (kidney stones). J. Mol. Med. 1997, 75, 160–163.
- Estévez, R.; Jentsch, T.J. CLC chloride channels: Correlating structure with function. Curr. Opin. Struct. Biol. 2002, 12, 531–539.
- Meyer, S.; Dutzler, R. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure 2006, 14, 299–307.
- Meyer, S.; Savaresi, S.; Forster, I.C.; Dutzler, R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 2007, 14, 60–67.
- Markovic, S.; Dutzler, R. The structure of the cytoplasmic domain of the chloride channel ClC-Ka reveals a conserved interaction interface. Structure 2007, 15, 715–725.
- Bateman, A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997, 22, 12–13.
- Schrecker, M.; Korobenko, J.; Hite, R.K. Cryo-EM structure of the lysosomal chloride-proton exchanger CLC-7 in complex with OSTM1. eLife 2020, 9, e59555.
- Park, E.; Campbell, E.B.; MacKinnon, R. Structure of a CLC chloride ion channel by cryo-electron microscopy. Nature 2017, 541, 500–505.
- Park, E.; MacKinnon, R. Structure of the CLC-1 chloride channel from Homo sapiens. eLife 2018, 7, e36629.
- Wang, K.; Preisler, S.S.; Zhang, L.; Cui, Y.; Missel, J.W.; Gronberg, C.; Gotfryd, K.; Lindahl, E.; Andersson, M.; Calloe, K.; et al. Structure of the human ClC-1 chloride channel. PLoS Biol. 2019, 17, e3000218.
- Pusch, M.; Ludewig, U.; Rehfeldt, A.; Jentsch, T.J. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature 1995, 373, 527–531.
- Traverso, S.; Elia, L.; Pusch, M. Gating competence of constitutively open CLC-0 mutants revealed by the interaction with a small organic Inhibitor. J. Gen. Physiol. 2003, 122, 295–306.
- Zifarelli, G.; Murgia, A.R.; Soliani, P.; Pusch, M. Intracellular proton regulation of ClC-0. J. Gen. Physiol. 2008, 132, 185–198.
- Estévez, R.; Boettger, T.; Stein, V.; Birkenhäger, R.; Otto, E.; Hildebrandt, F.; Jentsch, T.J. Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 2001, 414, 558–561.
- Jeworutzki, E.; López-Hernández, T.; Capdevila-Nortes, X.; Sirisi, S.; Bengtsson, L.; Montolio, M.; Zifarelli, G.; Arnedo, T.; Müller, C.S.; Schulte, U.; et al. GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl− channel auxiliary subunit. Neuron 2012, 73, 951–961.
- Jeworutzki, E.; Lagostena, L.; Elorza-Vidal, X.; Lopez-Hernandez, T.; Estevez, R.; Pusch, M. GlialCAM, a CLC-2 Cl− channel subunit, activates the slow gate of CLC chloride channels. Biophys. J. 2014, 107, 1105–1116.
- Lange, P.F.; Wartosch, L.; Jentsch, T.J.; Fuhrmann, J.C. ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature 2006, 440, 220–223.
- Stauber, T.; Weinert, S.; Jentsch, T.J. Cell biology and physiology of CLC chloride channels and transporters. Compr. Physiol. 2012, 2, 1701–1744.
- Bose, S.; He, H.; Stauber, T. Neurodegeneration upon dysfunction of endosomal/lysosomal CLC chloride transporters. Front. Cell Dev. Biol. 2021, 9, 639231.
- Günther, W.; Lüchow, A.; Cluzeaud, F.; Vandewalle, A.; Jentsch, T.J. ClC-5, the chloride channel mutated in Dent’s disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. USA 1998, 95, 8075–8080.
- Günther, W.; Piwon, N.; Jentsch, T.J. The ClC-5 chloride channel knock-out mouse—An animal model for Dent’s disease. Pflügers Arch. 2003, 445, 456–462.
- Jentsch, T.J.; Günther, W. Chloride channels: An emerging molecular picture. Bioessays 1997, 19, 117–126.
- Piwon, N.; Günther, W.; Schwake, M.; Bösl, M.R.; Jentsch, T.J. ClC-5 Cl−-channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature 2000, 408, 369–373.
- Stobrawa, S.M.; Breiderhoff, T.; Takamori, S.; Engel, D.; Schweizer, M.; Zdebik, A.A.; Bösl, M.R.; Ruether, K.; Jahn, H.; Draguhn, A.; et al. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 2001, 29, 185–196.
- Weinert, S.; Jabs, S.; Supanchart, C.; Schweizer, M.; Gimber, N.; Richter, M.; Rademann, J.; Stauber, T.; Kornak, U.; Jentsch, T.J. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl− accumulation. Science 2010, 328, 1401–1403.
- Ishida, Y.; Nayak, S.; Mindell, J.A.; Grabe, M. A model of lysosomal pH regulation. J. Gen. Physiol. 2013, 141, 705–720.
- Lloyd, S.E.; Pearce, S.H.; Fisher, S.E.; Steinmeyer, K.; Schwappach, B.; Scheinman, S.J.; Harding, B.; Bolino, A.; Devoto, M.; Goodyer, P.; et al. A common molecular basis for three inherited kidney stone diseases. Nature 1996, 379, 445–449.
- Kornak, U.; Kasper, D.; Bösl, M.R.; Kaiser, E.; Schweizer, M.; Schulz, A.; Friedrich, W.; Delling, G.; Jentsch, T.J. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001, 104, 205–215.
- Duncan, A.R.; Polovitskaya, M.M.; Gaitan-Penas, H.; Bertelli, S.; VanNoy, G.E.; Grant, P.E.; O’Donnell-Luria, A.; Valivullah, Z.; Lovgren, A.K.; England, E.M.; et al. Unique variants in CLCN3, encoding an endosomal anion/proton exchanger, underlie a spectrum of neurodevelopmental disorders. Am. J. Hum. Genet. 2021, 108, 1450–1465.
- Hu, H.; Haas, S.A.; Chelly, J.; Van Esch, H.; Raynaud, M.; de Brouwer, A.P.; Weinert, S.; Froyen, G.; Frints, S.G.; Laumonnier, F.; et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 2016, 21, 133–148.
- Palmer, E.E.; Pusch, M.; Picollo, A.; Forwood, C.; Nguyen, M.H.; Suckow, V.; Gibbons, J.; Hoff, A.; Sigfrid, L.; Megarbane, A.; et al. Functional and clinical studies reveal pathophysiological complexity of CLCN4-related neurodevelopmental condition. Mol. Psychiatry 2023, 28, 668–697.
- Palmer, E.E.; Stuhlmann, T.; Weinert, S.; Haan, E.; Van Esch, H.; Holvoet, M.; Boyle, J.; Leffler, M.; Raynaud, M.; Moraine, C.; et al. De novo and inherited mutations in the X-linked gene CLCN4 are associated with syndromic intellectual disability and behavior and seizure disorders in males and females. Mol. Psychiatry 2016, 23, 222–230.
- Polovitskaya, M.M.; Barbini, C.; Martinelli, D.; Harms, F.L.; Cole, F.S.; Calligari, P.; Bocchinfuso, G.; Stella, L.; Ciolfi, A.; Niceta, M.; et al. A Recurrent Gain-of-Function Mutation in CLCN6, Encoding the ClC-6 Cl−/H+-Exchanger, Causes Early-Onset Neurodegeneration. Am. J. Hum. Genet. 2020, 107, 1062–1077.
- Poët, M.; Kornak, U.; Schweizer, M.; Zdebik, A.A.; Scheel, O.; Hoelter, S.; Wurst, W.; Schmitt, A.; Fuhrmann, J.C.; Planells-Cases, R.; et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc. Natl. Acad. Sci. USA 2006, 103, 13854–13859.
- Zifarelli, G.; Pusch, M.; Fong, P. Altered voltage-dependence of slowly activating chloride-proton antiport by late endosomal ClC-6 explains distinct neurological disorders. J. Physiol. 2022, 600, 2147–2164.
- He, H.; Cao, X.; Yin, F.; Wu, T.; Stauber, T.; Peng, J. West syndrome caused by a chloride/proton exchange-uncoupling CLCN6 mutation related to autophagic-lysosomal dysfunction. Mol. Neurobiol. 2021, 58, 2990–2999.
- Kasper, D.; Planells-Cases, R.; Fuhrmann, J.C.; Scheel, O.; Zeitz, O.; Ruether, K.; Schmitt, A.; Poët, M.; Steinfeld, R.; Schweizer, M.; et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005, 24, 1079–1091.
- Pangrazio, A.; Pusch, M.; Caldana, E.; Frattini, A.; Lanino, E.; Tamhankar, P.M.; Phadke, S.; Lopez, A.G.; Orchard, P.; Mihci, E.; et al. Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: Report of 20 novel mutations. Hum. Mutat. 2010, 31, E1071–E1080.
- Nicoli, E.R.; Weston, M.R.; Hackbarth, M.; Becerril, A.; Larson, A.; Zein, W.M.; Baker, P.R., 2nd; Burke, J.D.; Dorward, H.; Davids, M.; et al. Lysosomal storage and albinism due to effects of a de novo CLCN7 variant on lysosomal acidification. Am. J. Hum. Genet. 2019, 104, 1127–1138.
- Steinmeyer, K.; Schwappach, B.; Bens, M.; Vandewalle, A.; Jentsch, T.J. Cloning and functional expression of rat CLC-5, a chloride channel related to kidney disease. J. Biol. Chem. 1995, 270, 31172–31177.
- Friedrich, T.; Breiderhoff, T.; Jentsch, T.J. Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J. Biol. Chem. 1999, 274, 896–902.
- Guzman, R.E.; Grieschat, M.; Fahlke, C.; Alekov, A.K. ClC-3 is an intracellular chloride/proton exchanger with large voltage-dependent nonlinear capacitance. ACS Chem. Neurosci. 2013, 4, 994–1003.
- Guzman, R.E.; Miranda-Laferte, E.; Franzen, A.; Fahlke, C. Neuronal ClC-3 splice variants differ in subcellular localizations, but mediate identical transport functions. J. Biol. Chem. 2015, 290, 25851–25862.
- Stauber, T.; Jentsch, T.J. Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J. Biol. Chem. 2010, 285, 34537–34548.
- Zifarelli, G.; Pusch, M. Conversion of the 2 Cl−/1 H+ antiporter ClC-5 in a NO3−/H+ antiporter by a single point mutation. Embo J. 2009, 28, 175–182.
- Brandt, S.; Jentsch, T.J. ClC-6 and ClC-7 are two novel broadly expressed members of the CLC chloride channel family. FEBS Lett. 1995, 377, 15–20.
- Ignoul, S.; Simaels, J.; Hermans, D.; Annaert, W.; Eggermont, J. Human ClC-6 is a late endosomal glycoprotein that associates with detergent-resistant lipid domains. PLoS ONE 2007, 2, e474.
- Neagoe, I.; Stauber, T.; Fidzinski, P.; Bergsdorf, E.Y.; Jentsch, T.J. The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J. Biol. Chem. 2010, 285, 21689–21697.
- Graves, A.R.; Curran, P.K.; Smith, C.L.; Mindell, J.A. The Cl−/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature 2008, 453, 788–792.
- Ludwig, C.F.; Ullrich, F.; Leisle, L.; Stauber, T.; Jentsch, T.J. Common gating of both CLC transporter subunits underlies voltage-dependent activation of the 2Cl−/1H+ exchanger ClC-7/Ostm1. J. Biol. Chem. 2013, 288, 28611–28619.
- Pusch, M.; Zifarelli, G. Large transient capacitive currents in wild-type lysosomal Cl−/H+ antiporter ClC-7 and residual transport activity in the proton glutamate mutant E312A. J. Gen. Physiol. 2021, 153, e202012583.
- Smith, A.J.; Lippiat, J.D. Voltage-dependent charge movement associated with activation of the CLC-5 2Cl−/1H+ exchanger. Faseb J. 2010, 24, 3696–3705.
- Zifarelli, G.; De Stefano, S.; Zanardi, I.; Pusch, M. On the mechanism of gating charge movement of ClC-5, a human Cl−/H+ antiporter. Biophys. J. 2012, 102, 2060–2069.
- Chalhoub, N.; Benachenhou, N.; Rajapurohitam, V.; Pata, M.; Ferron, M.; Frattini, A.; Villa, A.; Vacher, J. Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat. Med. 2003, 9, 399–406.



