Endosomes and lysosomes are intracellular vesicular organelles with important roles in cell functions such as protein homeostasis, clearance of extracellular material, and autophagy. Endolysosomes are characterized by an acidic luminal pH that is critical for proper function. Five members of the gene family of voltage-gated ChLoride Channels (CLC proteins) are localized to endolysosomal membranes, carrying out anion/proton exchange activity and thereby regulating pH and chloride concentration. Mutations in these vesicular CLCs cause global developmental delay, intellectual disability, various psychiatric conditions, lysosomal storage diseases, and neurodegeneration, resulting in severe pathologies or even death.
1. Introduction—The CLC Family
Physiologically, the most abundant anion is chloride. It is an important substrate of many transport proteins, being carried across the membrane as a single anion or coupled with other ions, and is important, for example, for the regulation of the membrane potential, intracellular vesicles acidification and cell volume regulation [
1].
In humans, the CLC family is formed by nine members, which had initially been supposed to be all chloride channels, because of their sequence homology with the founding member, the Torpedo electroplax channel ClC-0 [
2]. The discovery that the bacterial
Escherichia coli ecClC-1 homologue is not a passive chloride channel but a stoichiometrically coupled secondary active 2 Cl
−/1 H
+ antiporter has dramatically changed the point of view of the entire CLC group [
3]. Based on sequence homology, three branches of human CLCs have been distinguished. The first one includes the plasma membrane-localized chloride channels ClC-1, ClC-2 and the two isoforms ClC-Ka and ClC-Kb. The second branch is formed by ClC-3, ClC-4 and ClC-5, while the third branch contains ClC-6 and ClC-7. ClC-3 to -7 are all Cl
−/H
+ exchangers and are localized to the intracellular membranes of endosomes and/or lysosomes [
1].
All CLC family members share the same dimeric architecture that is unique to this protein family. Except for ClC-6 and ClC-7 [
4], the other CLC proteins can form homo- or hetero-dimers with members of the same branch [
1]. Biochemical studies and single-channel analysis on the first cloned
Torpedo ClC-0, mutants [
2,
5,
6] and biochemical and low-resolution structural analysis of ecClC-1 [
7,
8] suggested a homodimeric “double-barreled” architecture, with physically separated anion transport pathways in each protomer. This architecture has been fully confirmed by the determination of ecClC-1 and
Salmonella typhimurium stClC crystal structures [
9,
10]. The structures revealed the presence of distinct anion binding sites, formed by residues that are also highly conserved in human CLCs. The sites are denominated Sext, Scen and Sint, with Sext being occupied by the presumably negatively charged side chain of the “gating glutamate” E148 [
9,
10]. Each monomer presents 18 α-helices (from A to R) of which 17 are partially embedded in the membrane. The two subunits interact in a tight manner and the architecture follows an inverted and parallel orientation [
1]. Two C-terminal tandem cystathionine-β-synthase (CBS) domains are present in most eukaryotic CLC proteins [
11,
12], but are absent in ecClC-1. The two CBS domains may have a role in the so-called common gating process (that will be discussed in more detail below) and confer unique features to the CLC members [
1]. Dutzler and colleagues determined the crystal structures of isolated CBS domains of
Torpedo ClC-0 [
13], human ClC-5 [
14] and human ClC-Ka [
15]. CBS domains are present in many different protein families, where they are often implicated in the sensing of adenine nucleotides [
11,
16]. Structurally, so far, ATP has been found to be bound in the isolated domains of ClC-5 and in the full-length structure of ClC-7 [
17], but not in isolated domains of ClC-0 and ClC-Ka and not in full-length structures of bovine ClC-K or human ClC-1 [
18,
19,
20].
Single-channel recordings of the
Torpedo ClC-0 channel displayed two kinds of gating mechanisms that regulate the open probability (P
o) of the channel: a “
fast” or “
protopore” gate that acts independently on single pores determines the closing or opening state of each pore of the double-barreled structure [
1]. The fast gate is mainly determined by the gating glutamate (E166 in ClC-0), in that its neutralization renders CLC-0 channels voltage independent. Protonation of the gating glutamate and its competition with permeant ions underlie the anion and pH dependent protopore gating of most CLC channels [
10,
21,
22,
23]. Conversely, a second mechanism, termed a
slow or
common gate, operates on both pores simultaneously and is still not well understood [
1].
Some CLC proteins require association with a small ancillary subunit for proper function or membrane expression. In particular, the kidney ClC-Ka and ClC-Kb channels require association with the barttin subunit [
24]. In glia cells, ClC-2 associates with GlialCAM, a protein with the typical architecture of a cell adhesion molecule, which is mutated in megalencephalic leukoencephalopathy with subcortical cysts (MLC) [
25]. It leads to clustering of ClC-2 at glial cell–cell contacts and alters biophysical functions of the ClC-2 channel [
25,
26]. The complex of ClC-7 with its subunit Ostm1 is mandatory for mutual stabilization [
4,
27].
The plasma membrane localized chloride channels belonging to the first branch of the CLC family are expressed in a tissue-dependent manner that is different for each member according to their physiological role. All channel CLCs are involved in various human genetic diseases, as reviewed in detail elsewhere [
1,
28,
29].
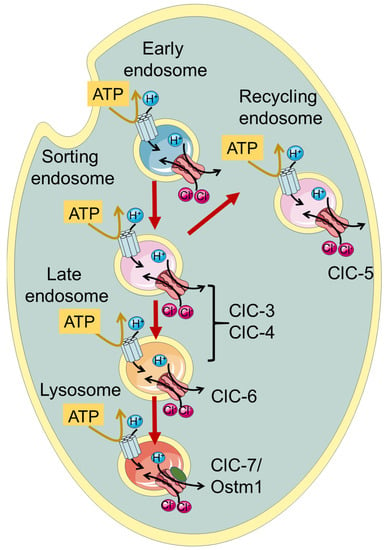
ClC-3 through ClC-7, which are the focus of this review, function as Cl
−/H
+ exchangers and are localized to intracellular endosomes and/or lysosomes (
Figure 1,
Table 1). Initially, when the transporter function of the intracellular CLCs was not yet known, it was proposed that they act as charge-shunting chloride channels to assist the luminal acidification of endosomes and lysosomes intracellular organelles [
30,
31,
32,
33,
34]. Indeed, the maintenance of an acidic pH of the lumen of endo-/lysosomes is required for their proper physiological function. The proton pumping V-ATPase is electrogenic and thus generates an electrical potential difference that would impede acidification if not neutralized by anionic cotransport and/or cationic counter-transport. Somewhat surprisingly and counter-intuitively, model calculations show that a 2 Cl
−/H
+ exchange activity, contributing to a more inside-negative voltage, allows a more acidic steady-state luminal pH compared to a shunting Cl
− channel [
35,
36].
Figure 1. Schematic illustration of localization of vesicular CLCs in the endo-/lysosomal pathway.
Among the endo-/lysosomal CLCs, ClC-5 is rather specifically expressed in the kidney with a predominant presence in epithelial cells of the proximal tubule, where it is involved in endocytic uptake [
30,
31,
33]. Indeed, mutations causing impaired ClC-5 transport activity are associated with Dent’s disease, a kidney disorder characterized by the primary symptom of low molecular weight proteinuria, and a series of secondary symptoms including kidney stones and renal failure, caused by defective endocytosis in the proximal tubule [
33,
37]. ClC-7, together with its subunit Ostm1 [
27], is rather ubiquitously expressed in the body and is localized to lysosomes and in the ruffled border of osteoclasts functioning as a 2Cl
−/H
+ antiporter [
1]. Accordingly, impaired bone resorption in osteoclast, caused by a functionally defective ClC-7/Ostm1 complex, causes osteopetrosis, a disease characterized by stiff and fragile bones [
38].
A large phenotypic spectrum of neuronal diseases is associated with mutations in the genes encoding ClC-3/-4/-6 and ClC-7, as will be described in detail in the following paragraphs (see Table 1).
For all vesicular CLCs, an unsolved question pertains to the direction of exchanger transport. Despite being physiologically localized to endo-/lysosomes, ClC-3 to -6 can reach the plasma membrane when heterologously expressed in HEK293 cells, allowing the investigation of their biophysical properties using the patch clamp technique [
45,
50,
51,
52,
53]. For ClC-7, the elimination of N-terminal lysosomal targeting motifs leads to plasma membrane expression [
4,
54]. ClC-3 to -5 all exhibit extreme outward rectification of currents with very little or nonresolvable activation kinetics [
50,
51,
52,
53,
55]. This current direction corresponds to the transport of luminal Cl
− out of lysosomes with a parallel influx of cytosolic H
+. However, it remains unclear whether the direction of transport is physiologically relevant and whether CLC exchangers work synergistically with V-ATPase, contributing to luminal acidification.
Additionally, ClC-6 and ClC-7 exhibit strongly outwardly rectifying currents, which are, however, characterized by slow activation kinetics and measurable inward “tail” currents [
4,
45].
2. ClC-3 and ClC-4
The second branch of the CLC family comprises ClC-3, -4 and 5. These three endosomal transporters share high sequence similarity and have similar functional properties [
1,
29]. Among the human CLCs, they are the most similar to the
Escherichia coli ecClC-1 homologue. The renal-specific ClC-5 is found mostly in recycling endosomes and its physiological role will not be discussed in detail [
1]. ClC-3 and ClC-4 are localized to sorting endosomes, and ClC-3 is probably localized in late endosomes as well [
1,
29]. ClC-3 has also been proposed to play a role in synaptic vesicles. This is, however, still controversial and will not be discussed in detail here.
While ClC-4 KO mice have no overt phenotype, in 2013 and 2016, patients (mostly pediatric) with a range of neurodevelopmental and psychiatric complications have been described with X-linked CLCN4 variants [40,42,65] (see Table 1). In heterologous expression, these and some novel variants [66] showed variable loss of function effects. It is important to note that complete loss of ClC-4 protein leads to non-syndromic intellectual disability in males and no disease in heterozygous females. In contrast, de novo and inherited missense variants can lead to severe syndromic neurological disease in males as well as in females, suggesting a dominant effect. In a more recent study, a large number of CLCN4 families was investigated, describing a large spectrum of clinical phenotypes and studying > 50 missense variants in heterologous expression [67]. Novel biophysical mechanisms were discovered for new and already described variants. These included a toxic gain of function characterized by the presence of negative currents at acidic extracellular (luminal) pH, and a shift in the voltage dependence of gating to more positive voltages [67]. Both effects can be expected to exert dominant negative effects in ClC-3/ClC-4 heterodimers.
Almost simultaneously came the discovery of the first variants in CLCN3 that cause global developmental delay, intellectual disability and neurodevelopmental disorders [39] (Table 1). Detailed functional analysis revealed a toxic gain of function for two missense variants, similar to the above-described effects in some CLCN4 variants [39].
3. ClC-6
The third branch of the CLC family comprises ClC-6 and ClC-7, which both function as Cl
−/H
+ exchangers [
1,
45]. Even though the expression of ClC-6 mRNA appears to be ubiquitous in many tissues [
67], biochemical analysis detected native ClC-6 protein predominantly in neurons, where it localizes to late endosomes and partially lysosomes [
44]. For a long time, the biophysical profile of ClC-6 remained completely unknown. In the first attempts at heterologous expression, no currents attributable to ClC-6 could be detected [
67], possibly caused by the intracellular localization of most of the overexpressed protein [
68].
A subtype of lysosomal storage disease, referred to as neuronal ceroid lipofuscinosis (NCL), was observed in ClC-6
knockout mice presenting a mild phenotype with features of reduced pain sensitivity, probably due to strong accumulation of materials in axon initial segments, mild cognitive abnormalities and no impact on their span life [
44]. This evidence suggested that
CLCN6 variants could be involved in human NCL [
44]. Indeed, in a sample of 75 adult-onset variants, including late-onset forms of NLC and Kufs’ disease, two individuals were found to be heterozygous for
CLCN6 missense variants (V580M and T628R) [
44]. However, no functional analysis had been performed at the time of that study because the transporter had not been successfully functionally characterized.
Preliminary electrophysiological characterization was obtained when the N-terminus of ClC-6 tagged with GFP (GFP-ClC-6) was reported to enhance its cell surface localization [
69]. However, the reported currents were small and barely above background levels.
4. ClC-7
Belonging to the third mammalian CLC branch, ClC-7 shares 45% of sequence homology with ClC-6. It was cloned in parallel with ClC-6 in 1995 [
67], but could not be functionally analyzed for a long time. Intriguingly, ClC-7 is the only subcellular CLC member to be present almost exclusively in lysosomes [
44]. Moreover, it has also been found in the ruffled border of osteoclasts, where it participates in bone resorption [
38]. Unlike the other CLC transporters, ClC-7 requires association with a type I transmembrane protein, called Ostm1, for proper function and stability [
4,
27].
Similarly to ClC-6, no information about electrophysiological ClC-7 characterization has been available for a long time, due to its intracellular localization upon heterologous expression [
1]. Ion flux studies with isolated mouse lysosomes showed that ClC-7 is the dominant anion permeation pathway of lysosomal membranes and that it performs 2 Cl
−/1 H
+ antiport activity [
72]. A breakthrough was achieved by Stauber and Jentsch, who discovered the sorting motifs that mediate lysosomal targeting [
54]. In particular, they found that when four leucine residues localized in the N-terminal portion are changed to alanine, the transporter is at least partially targeted to the plasma membrane [
54]. Notably, Ostm1 follows ClC-7 in its expression location. ClC-7
PM, the ClC-7 variant in which the two dileucine motifs are mutated to alanine, elicited robust transmembrane, outwardly rectifying voltage-activated currents [
4]. Even though some electrophysiological properties of ClC-7 are similar to that of other vesicular CLCs, including the inhibitory effect of acidic pH, ClC-7 differs substantially from ClC-3 to -5. Most importantly, ClC-7
PM exhibits very slow activation kinetics in the seconds time range [
4]. This slow “gating” phenomenon is strictly linked to conformational changes in the proteins, where the interactions between transmembrane as well as cytoplasmic domains play a key role [
73]. In addition to the transport currents, Pusch and Zifarelli discovered that the transporter also exhibits rather large “transient” or “capacitive” currents that reflect charge rearrangements within the protein. These are most likely mediated by movements of the gating glutamate and chloride binding/unbinding events [
71]. Similar currents have been observed in ClC-5 and ClC-3 [
52,
74,
75]. The transient currents probably have no physiological role, but represent a biophysical feature that can be useful in deciphering molecular mechanisms of gating and transport. Interestingly, while in ClC-5, neutralization of the so-called proton glutamate completely abolished transport currents, leaving only transient currents [
74,
75], in ClC-7, residual transport currents were observed in the corresponding E312A mutant [
71].
The physiological role of ClC-7 remained unclear for a long time. The first insights were obtained with a mouse KO model that was characterized by severe osteopetrosis [
38]. The involvement of ClC-7 in bone resorption was confirmed by the presence of
CLCN7 mutations in a human patient with malignant osteopetrosis [
38]. Further evidence came from the identification of a spontaneous Ostm1 mutation to be associated with the onset of a severe osteopetrosis in
gray lethal mice presenting a fur color defect [
76]. In Clcn7
−/− mice, even though the number of osteoclasts was normal, their ability to reabsorb calcified bone was impaired [
38]. Interestingly, however, no impact on lysosomal acidification was observed, suggesting that the osteoclasts’ ability to acidify intracellular vesicles was preserved in Clcn7
−/− mice [
38]. The life span of the KO mice was limited to 6–7 weeks.
This entry is adapted from the peer-reviewed paper 10.3390/life13061317