Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pierluigi Scalia | -- | 2024 | 2023-06-12 16:50:43 | | | |
| 2 | Dean Liu | -6 word(s) | 2018 | 2023-06-13 03:16:19 | | | | |
| 3 | Dean Liu | -3 word(s) | 2015 | 2023-06-20 02:36:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Scalia, P.; Williams, S.J.; Fujita-Yamaguchi, Y. Human IGF2 Gene. Encyclopedia. Available online: https://encyclopedia.pub/entry/45461 (accessed on 25 July 2026).
Scalia P, Williams SJ, Fujita-Yamaguchi Y. Human IGF2 Gene. Encyclopedia. Available at: https://encyclopedia.pub/entry/45461. Accessed July 25, 2026.
Scalia, Pierluigi, Stephen J. Williams, Yoko Fujita-Yamaguchi. "Human IGF2 Gene" Encyclopedia, https://encyclopedia.pub/entry/45461 (accessed July 25, 2026).
Scalia, P., Williams, S.J., & Fujita-Yamaguchi, Y. (2023, June 12). Human IGF2 Gene. In Encyclopedia. https://encyclopedia.pub/entry/45461
Scalia, Pierluigi, et al. "Human IGF2 Gene." Encyclopedia. Web. 12 June, 2023.
Copy Citation
Regulation of the human IGF2 gene displays multiple layers of control, which secures a genetically and epigenetically predetermined gene expression pattern throughout embryonal growth and postnatal life. These predominantly nuclear regulatory mechanisms converge on the function of the IGF2-H19 gene cluster on Chromosome 11 and ultimately affect IGF2 gene expression. Deregulation of such control checkpoints leads to the enhancement of IGF2 gene transcription and/or transcript stabilization, ultimately leading to IGF-II peptide overproduction. This type of anomaly is responsible for the effects observed in terms of both abnormal fetal growth and increased cell proliferation, typically observed in pediatric overgrowth syndromes and cancer.
IGF2, insulin-like growth factor 2 gene
mRNA transcript
IGF-II, insulin-like growth factor-2 peptide
1. IGF2 Gene Regulation at the Promoter and Transcript Level: An Unexploited View
A significant amount of experimental work has been previously produced to address the epigenetic control of the IGF2 gene among various species, supported by its well-known imprinting associated with DNA methylation [1][2][3]. IGF2 gene epigenetic regulation has been studied both during mammalian development as well as in IGF2-overexpressing syndromes and cancer [4][5][6][7]. Nonetheless, review work specifically addressing human IGF2 gene regulation at the transcriptional level is missing throughout the literature. This relative lack of recent experimental studies on IGF2-specific transcription factors has occurred despite the established biological role of IGF2 promoter regulation towards IGF2 transcriptional activation. A number of findings have also addressed IGF2 transcript regulation via non-coding RNA products (further discussed herein). More recently, among the known IGF2 (gene) regulatory mechanisms, the role of a family of IGF2–mRNA binding proteins (also known as IMPs) has emerged. In light of the increasing number of post-translational mechanisms regulating IGF2 expression and biological function, especially in cancer, researchers believe that focusing on the control of IGF2 at the gene transcription and RNA transcript levels and separating clearly from the post-translational mechanisms is far more important.
2. The Human IGF2 Gene Structure
The human IGF2 gene occupies the 11p15.5 chromosomal locus, positioned between the insulin gene and the H19 gene, with which it establishes an imprinted gene cluster (NCBI Ref Seq NG_008849.1) [8] (Figure 1). The IGF2 gene is composed of 10 exons [9][10][11] whose expression is driven by five promoters (p0–p4), differentially activated from embryonal to postnatal life (see Table 2) [12][13][14]. The IGF2 gene product is a peptidic ligand (IGF-II), which plays a central role in embryonal growth in mammalians [15]. Furthermore, the role of secreted IGF-II autocrine and paracrine effects in tumorigenesis [15] and its growing role towards malignant feature maintenance are well documented (reviewed by Scalia et al. [16]). Interestingly, an alternatively expressed exonic region has been recently described as part of exon 6, and it appears to bear a role in diabetes predisposition (see Figure 1) [17]. For this reason, the understanding of IGF2 gene expression and transcriptional regulation bears intrinsic high biological and biomedical value. The IGF2 gene has been widely studied for its epigenetic parental (allele)-specific control. The established evidence demonstrates that in the majority of adult tissues, IGF2 is exclusively expressed by the paternal (methylated) allele due to its imprinting on the maternal (hypomethylated) allele, which is silenced as a result of its hypo-methylation status. In particular, the IGF2 promoter-specific differentially methylated regions (DMRs 0, 1, 2) partially overlap the IGF2 intronic and exonic sequences, along with the DMR known as “inter-genic- or IG-DMR” or Imprinting Center Region 1, ICR1. This region is located between the IGF2 and H19 genes coding regions and the IGF2 enhancer region downstream from H19, cumulatively establishing a phylogenetically conserved gene cluster acting as an epigenetic switch [18][19][20]. IG-DMR is an allele-dependent DMR (ICR1) containing the binding motif for the epigenetic master regulator CTCF, which, along with the PRC2 complex components (summarized in Table 1). The CTCF–PRC2 complex binds the maternal hypomethylated ICR and insulates the IGF2 promoters [21][22]. On the contrary, the paternal ICR1, being prevented from CTCF binding as a consequence of ICR1 methylation status, results in a fully receptive effect of the enhancer regions, thereby displaying the classic monoallelic expression of the human imprinted IGF2 locus. This is graphically summarized in Figure 2A. A parallel promoter activation pattern for IGF2 expression in fetal growth, compared to the postnatal and adult phases, includes the promoter usage switching from the imprinted “fetal” (p2–p4) and “placental” (p0) promoters to the adult (p1) promoter [8]. Indeed, both cumulative and recent findings display a more diversified landscape of IGF2 regulation, extending beyond the previously known abnormalities linked to either (a) epigenetic deregulation or (b) allelic (uniparental) disomy, both of which are described in IGF2 overgrowth syndromes [23].
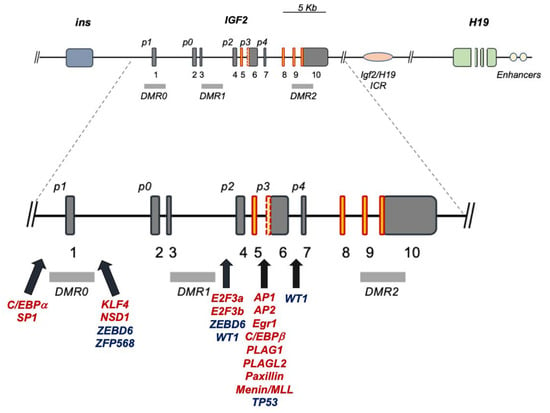
Figure 1. Human IGF2 gene structure and major transcriptional regulatory sites. Dark-grey boxes: non-coding exons; red-orange boxes: coding exons; red dotted yellow box between p3 and p4: alternatively expressed exon; p0–4: IGF2 promoter regions; light-grey rectangles: DMRs; red transcription factors (TFs): activators; blue TFs: repressors; black solid arrows: IGF2 promoter sites with cited TF binding motifs.
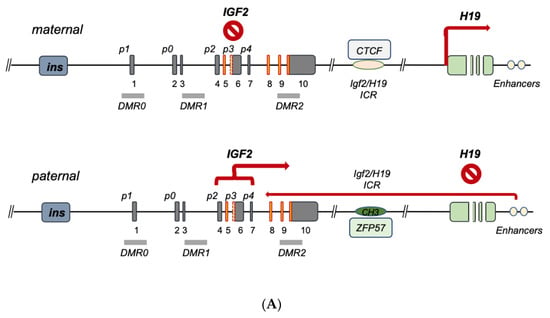
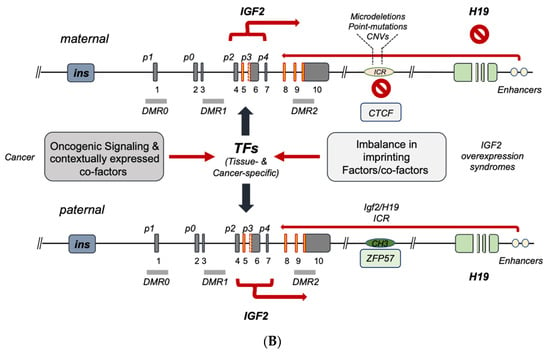
Figure 2. Current model for IGF2-H19 gene cluster regulation. (A) Imprinted IGF2 gene regulation in embryonal development, post-natal liver and tissues with maintenance of imprinting, MOI. The maternal allele bears a hypomethylated ICR1 allowing for CTFC binding; bound CTCF acts as an insulator by blocking the downstream enhancer effect on IGF2 which, as a result, is suppressed, while H19 transcription is unleashed by the same CTCF-ICR binding. On the paternal allele, ICR1 is methylated also through ZFP57, which contributes to the maintenance of the methylated status preventing CTCF binding; in absence of CTCF, the downstream enhancers trigger allele (paternal)-specific IGF2 transcription predominantly through its fetal promoters. (B) IGF2 gene regulation in cancer and IGF2 overexpression syndromes. In the case of IGF2 overexpression syndromes (e.g., Beckitt–Wiedemann), IGF2 expression abnormalities are mostly linked to a number of genetic abnormalities of the maternal IGF2-H19 ICR (microdeletions, CNVs, point mutations) leading to loss of CTCF binding, imprinting relaxation and biallelic expression of IGF2. In case of IGF2 regulation in cancers, a variety of regulatory modes have been described, which occur either independently or in parallel with the imprinting determining factors converging on each or both the maternal and paternal alleles and ultimately triggering the oncogenic activation of IGF2 transcription and/or the disruption of IGF2 transcriptional suppressors. The TF-mediated mechanisms conveyed in (B) relate to the potential full biallelic activation, but they can also be observed in monoallelic IGF2 overexpression.
3. IGF2 Gene Regulation during Development and IGF2 Overexpression Syndromes
Seminal studies have shown the importance of genomic imprinting for the IGF2 gene and the entire IGF2-H19 gene cluster (reviewed in [8]). As discussed in the previous section, ICR1 differentially methylated status affects the binding of epigenetic master-regulator CTCF to unmethylated ICR1 motifs acting as an insulator [2][21][24][25][26][27]. More recently, the role of the imprinting factor ZFP57 on the methylation maintenance status of the paternal allele has been shown [28]. The above control model for IGF2 imprinting on the maternal and paternal alleles is schematically summarized in Figure 2A.
The term ‘IGF2 overexpression syndromes’ relates to a variety of genetic abnormalities sharing the phenotype reported by Beckitt–Wiedemann to describe the resulting pediatric syndrome. A majority of the overgrowth symptoms in these subjects are secondary to the high levels of IGF-II produced at the embryonal and postnatal levels. This overproduction is mostly linked to the biallelic expression of IGF2 as a result of the imprinting relaxation of the maternal allele. A recent analysis of the genetic abnormalities in these subjects [23], leading to increased IGF2 activation, allows one to functionally classify the currently known IGF2 gene expression defects into two types, namely, (a) ICR defects causing the above relaxation on the maternal allele (via microdeletions and/or to DMR point mutations) [22][29][30][31], and (b) quantitative defects affecting the overall paternal gene cluster as a result of either uniparental disomy (UPD) or copy number variations (CNVs) [29][32][33]. An additional layer of control on IGF2 imprinting disclosed by recent studies relates to allele/DMR-specific factors acting as intrinsic-enabling factors and/or acting in synergy with CTCF on the maternal ICR [34]. Among these are Sox2/Oct4 [35][36], SUZ12 [37] and Vigilin [38], whose contextual functions as imprinting factors are governed by histone post-translational modifications, as evidenced by studies confirming their role in affecting both general and specific IGF2 imprinting. In particular, histone acetylation has been recognized since the late 1990s as a regulator of IGF2 imprinting, as shown by the ability of histone deacetylase inhibitors to cause IGF2 biallelic expression [39]. Even more detailed is the demonstration of the key role of H3K27 histone methylation for the proper maintenance of the maternal imprinting status via its effects on (a) the IGF2-H19 cluster loop conformation and (b) the DNA protein complex formation on the imprinted maternal allele [40]. In fact, in those cells with loss of imprinting (LOI), H3K27 demethylation leads to loss of the CTCF-orchestrated intrachromosomal loop between the IGF2 promoters and the ICR. The H3K27 methylation-free IGF2 promoters appear to become activated similarly to the paternal promoters, leading to biallelic expression. Noteworthily, SUZ12 has been shown to play a key role in the maintenance of the hypermethylation status of H3K27 by EZH2 since, in the absence of SUZ12, the PRC2 cannot be recruited to the maternal IGF2 promoter where this methylation takes place in order to induce the imprinting loop conformation [40]. The chromatin conformation at the IGF2-H19 cluster locus has been found to be essential for proper IGF2 expression, and this higher-order chromatin organization function is mediated by Cohesin [41]. Altogether, these studies point at a wider molecular network for the allele-specific control of IGF2 imprinting and offer additional potential mechanisms of dysregulation that could be responsible for those, yet unaccounted, molecular defects, leading to IGF2 increased transcription underlying the pathologic conditions discussed herein. A graphic summary of the human IGF2-H19 cluster regulation focusing on the latest landscape provided by the reviewed literature is conveyed in Figure 2 and Table 1.
Table 1. Factors binding IGF2-H19 ICR1 and affecting IGF2 imprinting status.
| Imprinting Factor | Key Feature | Reference(s) |
|---|---|---|
| CTCF | binds maternal ICR and insulates IGF2-p activity |
[34] |
| Cohesin | Cohesin is required for chromatin function at the H19/IGF2 locus | [41] |
| EZH2 | CH3-transferase component of PRC2 | [40] |
| SUZ12 | PRC2 component enabling ICR imprinting |
[37] |
| Sox2/Oct3–4 | CTCF-like effect | [35] |
| Vigilin | ICR imprinting effect via CTCF binding | [38] |
| ZFP57 | Binds paternal ICR and maintains methylated status | [28] |
4. IGF2 Gene Transcriptional Control in Cancer
A number of studies focusing on the role of IGF2 gene methylation and promoter usage in cancer have established the importance of IGF2 LOI status [42][43][44][45]. Nonetheless, the mechanistic relationship between promoter usage, both under monoallelic (under maintenance of imprinting, MOI) and biallelic status (caused by LOI), and the observed total IGF2 expression pattern/levels in cancer remains an active area of investigation. Indeed, a number of studies have shown a predominant activation of IGF2 fetal promoters (p2–p4) in a variety of cancers displaying IGF2 increased expression levels, with variable uncoupling of DMR0–2 methylation, along with monoallelic IGF2 and/or H19 expression [46][47][48][49][50][51][52][53][54][55]. In this context, it is important to stress that promoter usage and transcriptional activity are directly dependent on the involved transcription machinery, which is affected, in its turn, by the contextual transcriptional co-activator and co-repressor effects (also provided by the underlying IGF2 epigenetic protein–DNA interactions). In addition to the protein/DNA-driven control layer (or lack of control) of IGF2 gene expression, it is important to add the regulation layer provided by the RNA transcript control. This type of integrated approach to study IGF2 gene regulation both in IGF2 expression syndromes and in cancer, according to the authors of the present work, is essential in order to move the field beyond the historical (and still ongoing) compartmentalized approach to IGF2 gene studies. A graphic summary of the understanding of the regulation of the human IGF2 gene, spanning from IGF2 expression syndromes to cancer (overlapping in vivo), is provided in Figure 2B and Table 2.
Table 2. IGF2 promoter usage in physiology and disease.
| Promoter Usage | Imprinting Control | Reference(s) |
|---|---|---|
| IGF2-p0 | Not imprinted Mostly active in fetal placenta |
[14] |
| IGF2-p1 | Not imprinted—mostly active in postnatal Liver |
[56][57] |
| IGF2-p2 | Imprinted- Mostly active during Fetal growth |
[58] |
| IGF2-p3 & IGF2-p3/p4 (*) |
Imprinted- Mostly active during Fetal growth, Widely reactivated in cancer |
[50][56][57][59] |
(*) P3 and P4 are indicated together due to shared binding motifs often causing consensual activation.
References
- DeChiara, T.M.; Robertson, E.J.; Efstratiadis, A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 1991, 64, 849–859.
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485.
- Thorvaldsen, J.L.; Duran, K.L.; Bartolomei, M.S. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 1998, 12, 3693–3702.
- Sasaki, H.; Jones, P.A.; Chaillet, J.R.; Ferguson-Smith, A.C.; Barton, S.C.; Reik, W.; Surani, M.A. Parental imprinting: Potentially active chromatin of the repressed maternal allele of the mouse insulin-like growth factor II (Igf2) gene. Genes Dev. 1992, 6, 1843–1856.
- Morison, I.M.; Becroft, D.M.; Taniguchi, T.; Woods, C.G.; Reeve, A.E. Somatic overgrowth associated with overexpression of insulin-like growth factor II. Nat. Med. 1996, 2, 311–316.
- Ogawa, O.; Mishina, M.; Yoshida, O. Activation of imprinted genes in human carcinogenesis. Nihon rinsho. Jpn. J. Clin. Med. 1995, 53, 1009–1016.
- Taniguchi, T.; Sullivan, M.J.; Ogawa, O.; Reeve, A.E. Epigenetic changes encompassing the IGF2/H19 locus associated with relaxation of IGF2 imprinting and silencing of H19 in Wilms tumor. Proc. Natl. Acad. Sci. USA 1995, 92, 2159–2163.
- Reik, W.; Constancia, M.; Dean, W.; Davies, K.; Bowden, L.; Murrell, A.; Feil, R.; Walter, J.; Kelsey, G. Igf2 imprinting in development and disease. Int. J. Dev. Biol. 2000, 44, 145–150.
- Sussenbach, J.S.; Rodenburg, R.J.T.; Scheper, W.; Holthuizen, P. Transcriptional and Post-Transcriptional Regulation of the Human IGF-II Gene Expression. In Current Directions in Insulin-Like Growth Factor Research; Le Roith, D., Raizada, M.K., Eds.; Springer US: Boston, MA, USA, 1993; pp. 63–71.
- Mineo, R.; Fichera, E.; Liang, S.J.; Fujita-Yamaguchi, Y. Promoter usage for insulin-like growth factor-II in cancerous and benign human breast, prostate, and bladder tissues, and confirmation of a 10th exon. Biochem. Biophys. Res. Commun. 2000, 268, 886–892.
- Baral, K.; Rotwein, P. The insulin-like growth factor 2 gene in mammals: Organizational complexity within a conserved locus. PLoS ONE 2019, 14, e0219155.
- Holthuizen, P.; Van Dijk, M.A.; Rodenburg, R.J.; Koonen-Reemst, A.M.; Sussenbach, J.S. Transcriptional regulation of the major promoters of the human IGF-II gene. Mol. Reprod. Dev. 1993, 35, 391–393.
- Constancia, M.; Hemberger, M.; Hughes, J.; Dean, W.; Ferguson-Smith, A.; Fundele, R.; Stewart, F.; Kelsey, G.; Fowden, A.; Sibley, C.; et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002, 417, 945–948.
- Monk, D.; Sanches, R.; Arnaud, P.; Apostolidou, S.; Hills, F.A.; Abu-Amero, S.; Murrell, A.; Friess, H.; Reik, W.; Stanier, P.; et al. Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum. Mol. Genet. 2006, 15, 1259–1269.
- Christofori, G.; Naik, P.; Hanahan, D. A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature 1994, 369, 414–418.
- Scalia, P.; Giordano, A.; Williams, S.J. The IGF-II-Insulin Receptor Isoform-A Autocrine Signal in Cancer: Actionable Perspectives. Cancers 2020, 12, 366.
- Mercader, J.M.; Liao, R.G.; Bell, A.D.; Dymek, Z.; Estrada, K.; Tukiainen, T.; Huerta-Chagoya, A.; Moreno-Macias, H.; Jablonski, K.A.; Hanson, R.L.; et al. A Loss-of-Function Splice Acceptor Variant in IGF2 Is Protective for Type 2 Diabetes. Diabetes 2017, 66, 2903–2914.
- Davies, K.; Bowden, L.; Smith, P.; Dean, W.; Hill, D.; Furuumi, H.; Sasaki, H.; Cattanach, B.; Reik, W. Disruption of mesodermal enhancers for Igf2 in the minute mutant. Development 2002, 129, 1657–1668.
- Murrell, A.; Heeson, S.; Cooper, W.N.; Douglas, E.; Apostolidou, S.; Moore, G.E.; Maher, E.R.; Reik, W. An association between variants in the IGF2 gene and Beckwith-Wiedemann syndrome: Interaction between genotype and epigenotype. Hum. Mol. Genet. 2004, 13, 247–255.
- Charalambous, M.; Menheniott, T.R.; Bennett, W.R.; Kelly, S.M.; Dell, G.; Dandolo, L.; Ward, A. An enhancer element at the Igf2/H19 locus drives gene expression in both imprinted and non-imprinted tissues. Dev. Biol. 2004, 271, 488–497.
- Kurukuti, S.; Tiwari, V.K.; Tavoosidana, G.; Pugacheva, E.; Murrell, A.; Zhao, Z.; Lobanenkov, V.; Reik, W.; Ohlsson, R. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc. Natl. Acad. Sci. USA 2006, 103, 10684–10689.
- Murphy, S.K.; Erginer, E.; Huang, Z.; Visco, Z.; Hoyo, C. Genotype-Epigenotype Interaction at the IGF2 DMR. Genes 2015, 6, 777–789.
- Papulino, C.; Chianese, U.; Nicoletti, M.M.; Benedetti, R.; Altucci, L. Preclinical and Clinical Epigenetic-Based Reconsideration of Beckwith-Wiedemann Syndrome. Front. Genet. 2020, 11, 563718.
- Hark, A.T.; Schoenherr, C.J.; Katz, D.J.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000, 405, 486–489.
- Pant, V.; Mariano, P.; Kanduri, C.; Mattsson, A.; Lobanenkov, V.; Heuchel, R.; Ohlsson, R. The nucleotides responsible for the direct physical contact between the chromatin insulator protein CTCF and the H19 imprinting control region manifest parent of origin-specific long-distance insulation and methylation-free domains. Genes Dev. 2003, 17, 586–590.
- Lewis, A.; Murrell, A. Genomic imprinting: CTCF protects the boundaries. Curr. Biol. 2004, 14, R284–R286.
- Zhang, H.; Niu, B.; Hu, J.F.; Ge, S.; Wang, H.; Li, T.; Ling, J.; Steelman, B.N.; Qian, G.; Hoffman, A.R. Interruption of intrachromosomal looping by CCCTC binding factor decoy proteins abrogates genomic imprinting of human insulin-like growth factor II. J. Cell Biol. 2011, 193, 475–487.
- Riso, V.; Cammisa, M.; Kukreja, H.; Anvar, Z.; Verde, G.; Sparago, A.; Acurzio, B.; Lad, S.; Lonardo, E.; Sankar, A.; et al. ZFP57 maintains the parent-of-origin-specific expression of the imprinted genes and differentially affects non-imprinted targets in mouse embryonic stem cells. Nucleic Acids Res. 2016, 44, 8165–8178.
- Sparago, A.; Cerrato, F.; Vernucci, M.; Ferrero, G.B.; Silengo, M.C.; Riccio, A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat. Genet. 2004, 36, 958–960.
- Poole, R.L.; Leith, D.J.; Docherty, L.E.; Shmela, M.E.; Gicquel, C.; Splitt, M.; Temple, I.K.; Mackay, D.J. Beckwith-Wiedemann syndrome caused by maternally inherited mutation of an OCT-binding motif in the IGF2/H19-imprinting control region, ICR1. Eur. J. Hum. Genet. 2012, 20, 240–243.
- Abi Habib, W.; Brioude, F.; Azzi, S.; Salem, J.; Das Neves, C.; Personnier, C.; Chantot-Bastaraud, S.; Keren, B.; Le Bouc, Y.; Harbison, M.D.; et al. 11p15 ICR1 Partial Deletions Associated with IGF2/H19 DMR Hypomethylation and Silver-Russell Syndrome. Hum. Mutat. 2017, 38, 105–111.
- Haruta, M.; Arai, Y.; Sugawara, W.; Watanabe, N.; Honda, S.; Ohshima, J.; Soejima, H.; Nakadate, H.; Okita, H.; Hata, J.; et al. Duplication of paternal IGF2 or loss of maternal IGF2 imprinting occurs in half of Wilms tumors with various structural WT1 abnormalities. Genes Chromosomes Cancer 2008, 47, 712–727.
- Hubertus, J.; Lacher, M.; Rottenkolber, M.; Müller-Höcker, J.; Berger, M.; Stehr, M.; von Schweinitz, D.; Kappler, R. Altered expression of imprinted genes in Wilms tumors. Oncol. Rep. 2011, 25, 817–823.
- Li, T.; Hu, J.F.; Qiu, X.; Ling, J.; Chen, H.; Wang, S.; Hou, A.; Vu, T.H.; Hoffman, A.R. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol. Cell. Biol. 2008, 28, 6473–6482.
- Hori, N.; Yamane, M.; Kouno, K.; Sato, K. Induction of DNA demethylation depending on two sets of Sox2 and adjacent Oct3/4 binding sites (Sox-Oct motifs) within the mouse H19/insulin-like growth factor 2 (Igf2) imprinted control region. J. Biol. Chem. 2012, 287, 44006–44016.
- Higashimoto, K.; Jozaki, K.; Kosho, T.; Matsubara, K.; Fuke, T.; Yamada, D.; Yatsuki, H.; Maeda, T.; Ohtsuka, Y.; Nishioka, K.; et al. A novel de novo point mutation of the OCT-binding site in the IGF2/H19-imprinting control region in a Beckwith-Wiedemann syndrome patient. Clin. Genet. 2014, 86, 539–544.
- Wang, H.; Ge, S.; Qian, G.; Li, W.; Cui, J.; Wang, G.; Hoffman, A.R.; Hu, J.F. Restoration of IGF2 imprinting by polycomb repressive complex 2 docking factor SUZ12 in colon cancer cells. Exp. Cell Res. 2015, 338, 214–221.
- Liu, Q.; Yang, B.; Xie, X.; Wei, L.; Liu, W.; Yang, W.; Ge, Y.; Zhu, Q.; Zhang, J.; Jiang, L.; et al. Vigilin interacts with CCCTC-binding factor (CTCF) and is involved in CTCF-dependent regulation of the imprinted genes Igf2 and H19. FEBS J. 2014, 281, 2713–2725.
- Hu, J.F.; Oruganti, H.; Vu, T.H.; Hoffman, A.R. The role of histone acetylation in the allelic expression of the imprinted human insulin-like growth factor II gene. Biochem. Biophys. Res. Commun. 1998, 251, 403–408.
- Li, T.; Chen, H.; Li, W.; Cui, J.; Wang, G.; Hu, X.; Hoffman, A.R.; Hu, J. Promoter histone H3K27 methylation in the control of IGF2 imprinting in human tumor cell lines. Hum. Mol. Genet. 2014, 23, 117–128.
- Nativio, R.; Wendt, K.S.; Ito, Y.; Huddleston, J.E.; Uribe-Lewis, S.; Woodfine, K.; Krueger, C.; Reik, W.; Peters, J.M.; Murrell, A. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009, 5, e1000739.
- Issa, J.P.; Vertino, P.M.; Boehm, C.D.; Newsham, I.F.; Baylin, S.B. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 11757–11762.
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755.
- Bjornsson, H.T.; Brown, L.J.; Fallin, M.D.; Rongione, M.A.; Bibikova, M.; Wickham, E.; Fan, J.B.; Feinberg, A.P. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J. Natl. Cancer Inst. 2007, 99, 1270–1273.
- Lu, L.; Cai, M.; Peng, M.; Wang, F.; Zhai, X. miR-491-5p functions as a tumor suppressor by targeting IGF2 in colorectal cancer. Cancer Manag. Res. 2019, 11, 1805–1816.
- Li, X.; Adam, G.; Cui, H.; Sandstedt, B.; Ohlsson, R.; Ekstrom, T.J. Expression, promoter usage and parental imprinting status of insulin-like growth factor II (IGF2) in human hepatoblastoma: Uncoupling of IGF2 and H19 imprinting. Oncogene 1995, 11, 221–229.
- Douc-Rasy, S.; Barrois, M.; Fogel, S.; Ahomadegbe, J.C.; Stéhelin, D.; Coll, J.; Riou, G. High incidence of loss of heterozygosity and abnormal imprinting of H19 and IGF2 genes in invasive cervical carcinomas. Uncoupling of H19 and IGF2 expression and biallelic hypomethylation of H19. Oncogene 1996, 12, 423–430.
- Vu, T.H.; Hoffman, A. Alterations in the promoter-specific imprinting of the insulin-like growth factor-II gene in Wilms’ tumor. J. Biol. Chem. 1996, 271, 9014–9023.
- Hodzic, D.; Frey, B.; Marechal, D.; Scarcez, T.; Grooteclaes, M.; Winkler, R. Cloning of breakpoints in and downstream the IGF2 gene that are associated with overexpression of IGF2 transcripts in colorectal tumours. Oncogene 1999, 18, 4710–4717.
- Eriksson, T.; Frisk, T.; Gray, S.G.; von Schweinitz, D.; Pietsch, T.; Larsson, C.; Sandstedt, B.; Ekström, T.J. Methylation Changes in the Human IGF2 P3 Promoter Parallel IGF2 Expression in the Primary Tumor, Established Cell Line, and Xenograft of a Human Hepatoblastoma. Exp. Cell Res. 2001, 270, 88–95.
- Kim, S.J.; Park, S.E.; Lee, C.; Lee, S.Y.; Jo, J.H.; Kim, J.M.; Oh, Y.K. Alterations in promoter usage and expression levels of insulin-like growth factor-II and H19 genes in cervical carcinoma exhibiting biallelic expression of IGF-II. Biochim. Biophys. Acta 2002, 1586, 307–315.
- Yu, Y.; Wylie-Sears, J.; Boscolo, E.; Mulliken, J.B.; Bischoff, J. Genomic imprinting of IGF2 is maintained in infantile hemangioma despite its high level of expression. Mol. Med. 2004, 10, 117–123.
- Murphy, S.K.; Huang, Z.; Wen, Y.; Spillman, M.A.; Whitaker, R.S.; Simel, L.R.; Nichols, T.D.; Marks, J.R.; Berchuck, A. Frequent IGF2/H19 domain epigenetic alterations and elevated IGF2 expression in epithelial ovarian cancer. Mol. Cancer Res. 2006, 4, 283–292.
- Qian, B.; Katsaros, D.; Lu, L.; Canuto, E.M.; Benedetto, C.; Beeghly-Fadiel, A.; Yu, H. IGF-II promoter specific methylation and expression in epithelial ovarian cancer and their associations with disease characteristics. Oncol. Rep. 2011, 25, 203–213.
- Küffer, S.; Gutting, T.; Belharazem, D.; Sauer, C.; Michel, M.S.; Marx, A.; Trojan, L.; Ströbel, P. Insulin-like growth factor 2 expression in prostate cancer is regulated by promoter-specific methylation. Mol. Oncol. 2018, 12, 256–266.
- Nardone, G.; Romano, M.; Calabrò, A.; Pedone, P.V.; de Sio, I.; Persico, M.; Budillon, G.; Bruni, C.B.; Riccio, A.; Zarrilli, R. Activation of fetal promoters of insulin-like growth factors II gene in hepatitis C virus-related chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Hepatology 1996, 23, 1304–1312.
- Li, X.; Nong, Z.; Ekström, C.; Larsson, E.; Nordlinder, H.; Hofmann, W.J.; Trautwein, C.; Odenthal, M.; Dienes, H.P.; Ekström, T.J.; et al. Disrupted IGF2 promoter control by silencing of promoter P1 in human hepatocellular carcinoma. Cancer Res. 1997, 57, 2048–2054.
- Lui, J.C.; Baron, J. Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proc. Natl. Acad. Sci. USA 2013, 110, 6181–6186.
- Li, Y.; Meng, G.; Huang, L.; Guo, Q.N. Hypomethylation of the P3 promoter is associated with up-regulation of IGF2 expression in human osteosarcoma. Hum. Pathol. 2009, 40, 1441–1447.
More
Information
Subjects:
Genetics & Heredity
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
3 times
(View History)
Update Date:
20 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No