Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ondrej Fabian | -- | 2237 | 2023-06-08 14:29:45 | | | |
| 2 | Fanny Huang | Meta information modification | 2237 | 2023-06-12 02:56:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Fabian, O.; Bajer, L.; Drastich, P.; Harant, K.; Sticova, E.; Daskova, N.; Modos, I.; Tichanek, F.; Cahova, M. Proteomics in Adult and Pediatric Inflammatory Bowel Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/45349 (accessed on 25 July 2026).
Fabian O, Bajer L, Drastich P, Harant K, Sticova E, Daskova N, et al. Proteomics in Adult and Pediatric Inflammatory Bowel Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/45349. Accessed July 25, 2026.
Fabian, Ondrej, Lukas Bajer, Pavel Drastich, Karel Harant, Eva Sticova, Nikola Daskova, Istvan Modos, Filip Tichanek, Monika Cahova. "Proteomics in Adult and Pediatric Inflammatory Bowel Diseases" Encyclopedia, https://encyclopedia.pub/entry/45349 (accessed July 25, 2026).
Fabian, O., Bajer, L., Drastich, P., Harant, K., Sticova, E., Daskova, N., Modos, I., Tichanek, F., & Cahova, M. (2023, June 08). Proteomics in Adult and Pediatric Inflammatory Bowel Diseases. In Encyclopedia. https://encyclopedia.pub/entry/45349
Fabian, Ondrej, et al. "Proteomics in Adult and Pediatric Inflammatory Bowel Diseases." Encyclopedia. Web. 08 June, 2023.
Copy Citation
Inflammatory bowel diseases (IBD) are systemic immune-mediated conditions with predilection for the gastrointestinal tract and include Crohn’s disease and ulcerative colitis. Genomic and transcriptomic studies contributed substantially to our understanding of the immunopathological pathways involved in disease initiation and progression. However, eventual genomic alterations do not necessarily translate into the final clinical picture. Proteomics represent a missing link between the genome, transcriptome, and phenotypical presentation of the disease. Based on the analysis of a large spectrum of proteins in tissues, it seems to be a promising method for the identification of new biomarkers.
inflammatory bowel disease
pediatric
proteomics
proteome
ulcerative colitis
1. Introduction
Inflammatory bowel diseases (IBD) represent chronic systemic immune-mediated conditions with a predilection for the gastrointestinal (GI) tract and include Crohn’s disease (CD) and ulcerative colitis (UC) as two major phenotypes. Currently, IBD are perceived more likely as a continuous spectrum of disorders ranging from CD with a small bowel and/or upper GI predilection across colonic CD to IBD unclassified (IBDU) to distal UC [1][2]. The annual incidence of CD ranges between 0.3 and 12.7/100,000 persons and 0 and 20.2/100,000 in Europe and North America, respectively. For UC, the annual incidence is 0.6–24.3/100,000 in Europe and 0–19.2/100,000 in North America. The highest incidence worldwide can be seen in the Faroe Islands, reaching up to 81/100,000 inhabitants [3][4]. There is a well-known west–east and north–south descending gradient with the highest occurrence of the disease in countries in Northern and Western Europe and North America [5]. Although the disease may manifest at any age, young adults are the most commonly affected group. The second peak can be seen in older patients around their seventh decade. Pediatric IBD represent approximately 1/4 of all cases [6][7]. The etiology of the disease remains largely unknown, and the pathogenesis is poorly understood. However, the impaired homeostasis between the intestinal epithelial barrier, immune barrier, and commensal intestinal microbiota in a genetically susceptible host is considered to be a key mechanism of the disease’s initiation and progression (Figure 1) [8].
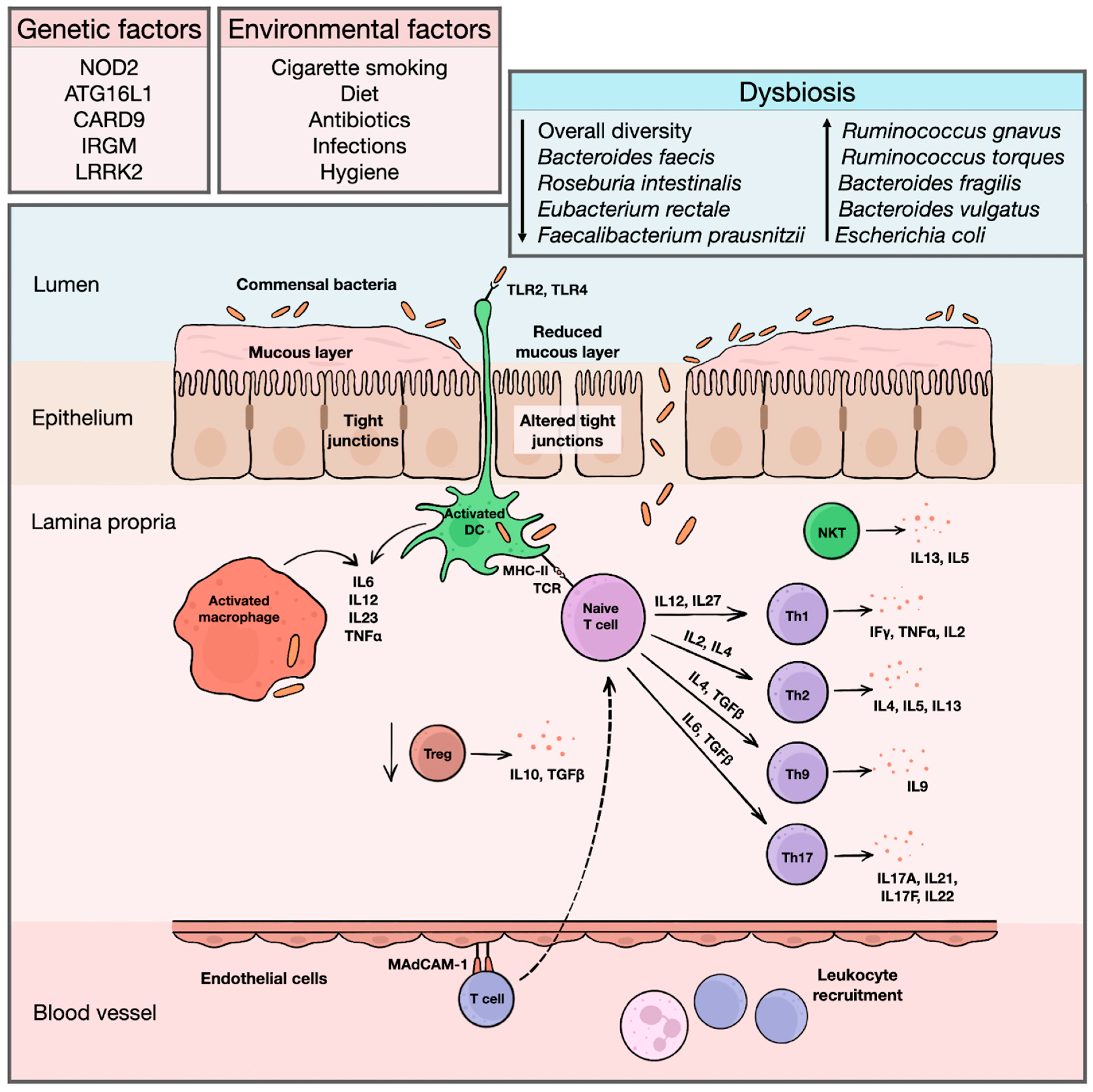
Figure 1. An overview of IBD etiopathogenesis. Exact etiopathogenic mechanisms of IBD initiation and progression are still largely unknown. However, the etiology of the diseases is probably multifactorial, with a contribution of both genetic and environmental factors altering physiological composition of intestinal microbiota and initiating an inadequate inflammatory response. Bacterial antigens are recognized by toll-like receptors on surface of dendritic antigen-presenting cells, macrophages and intestinal epithelia. In healthy individuals, antigen-presenting cells induce immune tolerance and stimulate differentiation of regulatory T cells, producing anti-inflammatory cytokines, such as IL10 or TGFβ. In IBD, changes in intestinal microbiota, together with increased permeability of intestinal epithelium and reduced luminal mucous layer facilitating translocation of bacterial fragments into lamina propria, triggers inflammatory response. Activated dendritic cells and macrophages produce various pro-inflammatory cytokines, such as IL2 or IL23, and initiate differentiation of naïve T cells into effector T cells. For a long time, Crohn’s disease was considered a disease driven predominantly by Th1 cells with a production of high amounts of IFNγ, TNFα, or IL2, while an ulcerative colitis was linked with Th2-predominant immune pathway and secretion of IL4, IL5, or IL13. In both diseases, there is also a strong influence of Th17-related cytokines, such as IL17A or IL17F. Apart from that, a substantial contribution of Th9 cells and natural killer T cells is recognized, further disrupting the intestinal epithelial integrity. Simultaneously, additional leukocytes are recruited from peripheral circulation. These cells bind to endothelial cells lining the mucosal microvasculature via mucosal vascular addressin–cell adhesion molecule 1, whose expression is upregulated in inflamed mucosa, and migrate to affected area of lamina propria, maintaining chronic inflammatory reaction. ATG16L1 = autophagy-related 16-like 1 gene; CARD9 = caspase recruitment domain-containing protein 9 gene; DC = dendritic cell; IBD = inflammatory bowel diseases; IFγ = interferon gamma; IL = interleukin; IRGM = immunity-related GTPase family M protein gene; LRRK2 = leucine-rich repeat kinase 2 gene; MAdCAM-1 = mucosal vascular addressin–cell adhesion molecule 1; MHC-II = major histocompatibility complex class II; NKT = natural killer T cell; NOD2 = nucleotide binding oligomerization domain containing 2 gene; TCR = T cell receptor; TGFβ = transforming growth factor beta; Th = helper T cell; TLR = toll-like receptor; TNFα = tumor necrosis factor alpha; Treg = regulatory T cell.
The advances in the diagnostics and implementation of modern therapeutic approaches improved the overall clinical management of patients. However, up to 6% of adult and 13% of pediatric IBD cases remain further unclassified and many patients still face an adverse clinical course with frequent recurrences or severe complications [7]. The variable clinical presentation and uneven response to therapy suggest that etiopathogenic mechanisms of the disease progression are probably prone to substantial inter-individual variability. There is, thus, demand for new invasive or non-invasive biomarkers that would facilitate a diagnostic process, reflecting the actual intensity of inflammation and predicting the subsequent clinical course or development of complications.
A biomarker is defined as a measurable indicator of a specific biological state [9]. Many promising serum, fecal, or tissue biomarkers were introduced over time, but only a small fraction was implemented in routine clinical practice. The ideal biomarker should be non-invasive, highly sensitive and specific, easily detectable, and financially reasonable. To this day, no such biomarker has been identified. Proteomics represents a promising method of identification of potential new biomarkers based on the analysis of a large spectrum of proteins from tissue samples or body fluids.
2. The Value of Proteomics
The term proteome describes a full set of proteins translated from a transcriptome within a single cell, tissue, or specific cellular compartment [10][11]. Proteomics then refers to a large-scale study analyzing a proteome. In practice, it is understood as a research sphere aiming at the identification of the largest number of proteins or peptides possible in the given tissue, clarifying their functions and understanding their reciprocal interactions [12]. It is well known that the number of specific protein variants in our tissues greatly exceeds the number of protein-coding genes [13]. Currently, more than 20,000 such genes are known. Each gene may give rise to several different RNA transcripts, which are subsequently translated to proteins. The proteins themselves may undergo a series of post-translational modifications, such as phosphorylation, glycosylation, or methylation. Currently, several such modifications are recognized. Each protein, thus, may give rise to hundreds of specific proteoforms [13][14]. Many additional arrangements throughout the transcriptomic or translational process also play a role, such as the regulation of translational rate or diversity of protein transport [13][14][15]. It is estimated that hundreds of thousands or maybe even millions of specific proteoforms exist in the human body [14][16]. It is, thus, obvious that a spectrum of potential proteins involved in the specific biological process cannot be reliably estimated solely via genomic or transcriptomic analysis.
In IBD, a genetic background plays an important role in the etiopathogenesis of the disease. Advances in molecular genetics and the introduction of genome-wide association studies allowed the detection of many candidate genes, whose pathogenic mutations or single nucleotide polymorphisms show an association with an increased risk of CD or UC. However, it is the proteomics that may finally link genomic and transcriptomic alterations with the phenotypical presentation of IBD and allow an understanding of mechanisms responsible for the initiation and progression of the disease. Previously, a search for candidate biomarkers was limited to a shortlist of proteins suspected to be involved in IBD pathogenesis. Advancements in modern proteomic techniques, such as mass spectrometry (MS) and implementation of complex bioinformatic analyses, enabled the isolation and analysis of a large number of proteins, increasing the chance of a novel biomarker discovery [17].
Standard proteomic research aiming at the identification of new potential biomarkers follows several phases, including biomarker discovery, verification, and validation [18]. The first phase begins with an analysis of a tissue, body fluid (i.e., serum), stool, or isolated cells, from which a large spectrum of proteins is identified. Those subjects with the biggest difference in expression between the cohorts are further examined and several candidate proteins are proposed. Since this phase is usually both financially and time demanding, studies are often performed on small cohorts of patients with a high risk of false negative or false positive findings [9][18]. The subsequent verification phase tests the sensitivity and specificity of the candidate biomarkers on larger cohorts of patients. Usually, antibody-based techniques, such as enzyme-linked immunosorbent assay (ELISA) or Western blot, are applied. The amount of candidate biomarkers is usually significantly reduced after this phase [9][17][18].
The newly described biomarkers can serve several clinical purposes. In the context of IBD, they can facilitate the following actions: (1) the distinguishing of IBD from other diseases; (2) the differentiation of IBD phenotypes; (3) the understanding of the pathogenic processes in the respective IBD subtypes; (4) the monitoring of the disease activity; (5) the prediction of severe clinical complications or a disease relapse; (6) the prediction of responsiveness to therapy, and (7) the prediction of neoplastic transformation.
3. Techniques in Proteomics
Proteomic studies usually follow one of the two main analytical approaches termed “top-down” and “bottom-up”. The bottom-up approach identifies proteins based on the analysis of small peptide fragments created via previous enzymatic proteolysis [19]. The top-down strategy, on the other hand, recognizes whole proteins that did not undergo previous enzymatic lysis [20]. The first approach is a commonly used strategy of protein identification but entails a risk of losing less abundant protein fragments, a higher percentage of peptide overlaps, and an inferior quality of post-translational modification analysis. The second approach has a lower sensitivity in the large-scale identification of proteins from complex mixtures and is prone to a higher dynamic range challenge in protein identification and characterization. However, it may be better suited for the analysis of proteins with multiple different proteoforms [21][22].
The most commonly used proteomic techniques can be subclassified into two main categories, termed “gel-based” and “gel-free”. Gel-based techniques include polyacrylamide gel electrophoresis (PAGE), two-dimensional difference gel electrophoresis (2-DE of 2D-PAGE), and difference gel electrophoresis (DIGE). These techniques separate proteins on the basis of their isoelectric point and, in the case of the two-dimensional approach, also based on the molecular mass. The individual protein groups are then represented by spots in the gel medium [23][24]. The spots subsequently undergo an in-gel digestion via endopeptidases or enzymatic digestion after their excision from the gel. As the next step, they are usually analyzed via MS [25]. However, gel-based methods are labor-intensive and limited in their ability to detect low-abundance proteins or proteins with a high molecular mass [26]. Gel-free methods are much more straightforward. Sample is homogenized and directly proteolytically cleaved. Resulting peptides are separated usually via reverse phase liquid chromatography (LC) and eluting peptides are detected with MS. LC is a technique able to separate peptides from digested cell lysates for a subsequent MS analysis on the basis of their chemical properties, mainly on hydrophobicity. The analyzed molecules interact with stationary phase attached to the surface of sorbent beads in LC column. Molecules are separated while moving through column with the aid of a mobile phase [27][28]. The most commonly used approach for peptide quantification is a label-free approach in data dependent (DDA) or data independent (DIA) acquisition mode. In DDA mode, each fragmentation spectrum originates from the fragmentation of one isolated precursor. On the other hand, in DIA mode, multiple precursors are fragmented together, and the resulting fragments are assigned to peptide precursors during data processing [29][30]. Alternatively, isobaric tags for relative quantification (iTRAQ) or stable isotope labelling with amino acids in cell culture (SILAC) can be used. iTRAQ is a method based on the covalent binding of the N-terminus and side chain amines of peptides with isobaric tags of varying mass [31]. A similar approach to iTRAQ is a tandem mass tag (TMT) that varies with regard to the chemical structure of the used tags [32]. SILAC is a method in which stable isotope-labeled amino acids are incorporated into the proteome. It is usually performed on cell mixtures containing two cell populations. One population is fed with a growth medium containing labeled amino acids. Proteins from both cell populations are then combined and analyzed together via a MS. The labeled and non-labeled amino acids have a known mass shift that allows the comparison of a given peptide between both samples. The ratio of peak intensities in the mass spectrum for such peptide pairs reflects the abundance ratio for the two proteins [33][34][35].
Regardless of the protein preparation technique, the process is usually followed by a MS analysis. The basic workflow can be summarized in three steps: (1) protein/peptide ionization, (2) separation of ions on the basis of their mass-to-charge ratio, and (3) ion detection. If the analysis follows a top-down approach, the samples are usually analyzed directly via a MS. In bottom-up strategy, the proteins are cleaved to peptides and consequently separated via LC or directly analyzed via a MS. The most common ionization sources include matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI). MALDI technique is usually preferred due to its capacity to produce singly charged ions of peptides, thus minimizing the complexity of the given spectra. The most frequently used ion analyzers are time-of-flight (TOF) or Fourier transform-based instruments (Orbitrap, FT-ICR-MS). In most instances, peptides undergo several rounds of fragmentation and MS analysis, termed tandem MS or MS/MS. In modern proteomics, ESI-based hybrid instruments are usually combined quadrupole with a TOF- or Orbitrap-based spectrometer. This technique is used to determine peptide sequences, while standard MS workflow is usually reserved for peptide mass measurements [36][37][38]. A flowchart showing the basic proteomic approaches is shown in Figure 2.
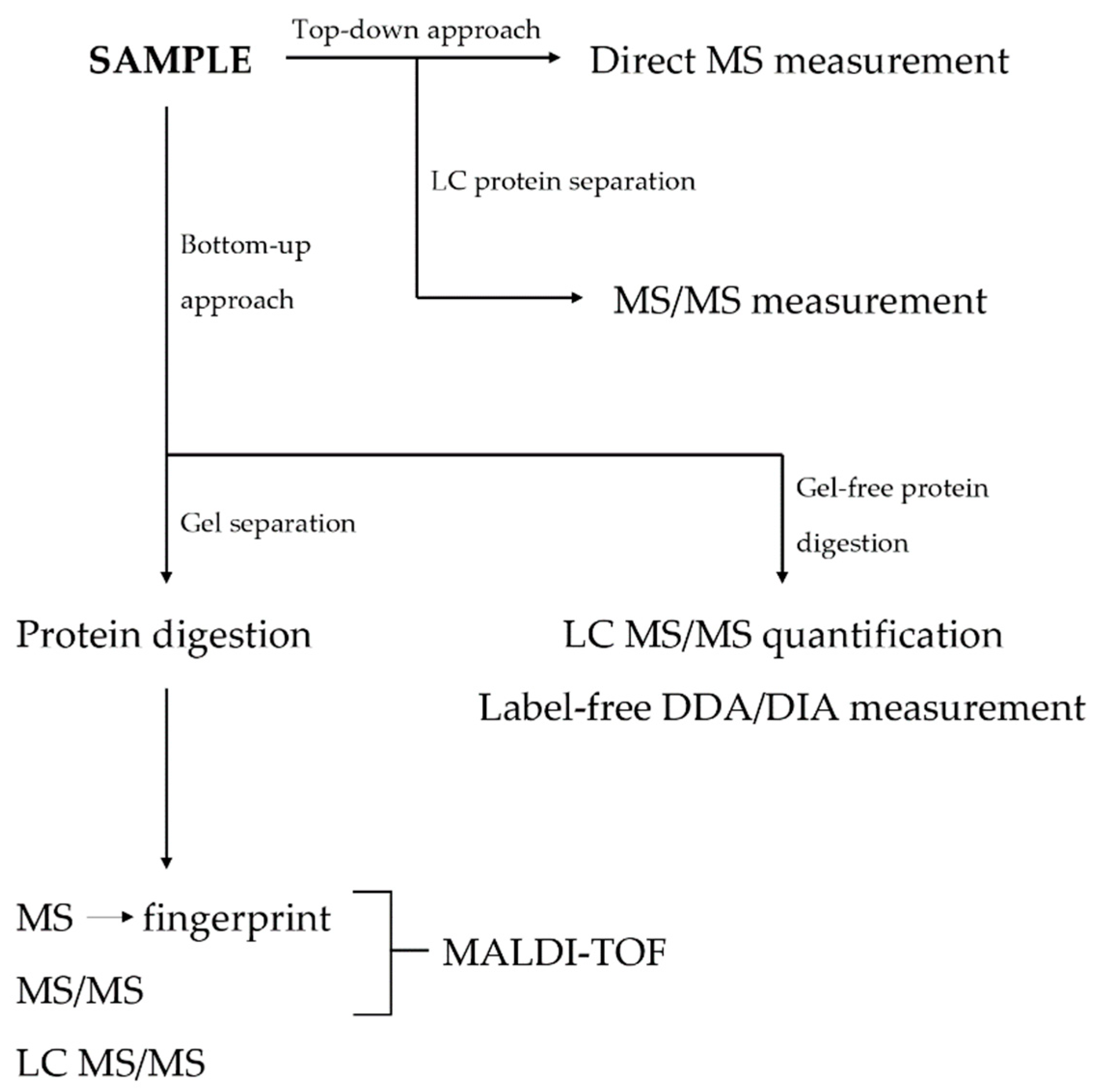
Figure 2. A flowchart of basic proteomic approaches. DDA = data-dependent acquisition; DIA = data-independent acquisition; LC = liquid chromatography; MALDI = matrix-assisted laser desorption/ionization; MS = mass spectrometry; MS/MS = tandem mass spectrometry; TOF = time of flight.
References
- Magro, F.; Langner, C.; Driessen, A.; Ensari, A.; Geboes, K.; Mantzaris, G.J.; Villanacci, V.; Becheanu, G.; Borralho Nunes, P.; Cathomas, G.; et al. European consensus on the histopathology of inflammatory bowel disease. J. Crohns Colitis 2013, 7, 827–851.
- Levine, A.; Koletzko, S.; Turner, D.; Escher, J.C.; Cucchiara, S.; de Ridder, L.; Kolho, K.L.; Veres, G.; Russell, R.K.; Paerregaard, A.; et al. ESPGHAN revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 795–806.
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42, quiz e30.
- Benchimol, E.I.; Fortinsky, K.J.; Gozdyra, P.; Van den Heuvel, M.; Van Limbergen, J.; Griffiths, A.M. Epidemiology of pediatric inflammatory bowel disease: A systematic review of international trends. Inflamm. Bowel Dis. 2011, 17, 423–439.
- Burisch, J.; Pedersen, N.; Cukovic-Cavka, S.; Brinar, M.; Kaimakliotis, I.; Duricova, D.; Shonova, O.; Vind, I.; Avnstrom, S.; Thorsgaard, N.; et al. East-West gradient in the incidence of inflammatory bowel disease in Europe: The ECCO-EpiCom inception cohort. Gut 2014, 63, 588–597.
- Conrad, M.A.; Rosh, J.R. Pediatric Inflammatory Bowel Disease. Pediatr. Clin. North Am. 2017, 64, 577–591.
- Prenzel, F.; Uhlig, H.H. Frequency of indeterminate colitis in children and adults with IBD—A metaanalysis. J. Crohns Colitis 2009, 3, 277–281.
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640.
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983.
- Tyers, M.; Mann, M. From genomics to proteomics. Nature 2003, 422, 193–197.
- Assadsangabi, A.; Evans, C.A.; Corfe, B.M.; Lobo, A. Application of Proteomics to Inflammatory Bowel Disease Research: Current Status and Future Perspectives. Gastroenterol. Res. Pract. 2019, 2019, 1426954.
- Baldan-Martin, M.; Chaparro, M.; Gisbert, J.P. Tissue Proteomic Approaches to Understand the Pathogenesis of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1184–1200.
- Bennike, T.; Birkelund, S.; Stensballe, A.; Andersen, V. Biomarkers in inflammatory bowel diseases: Current status and proteomics identification strategies. World J. Gastroenterol. 2014, 20, 3231–3244.
- Lawrance, I.C.; Klopcic, B.; Wasinger, V.C. Proteomics: An overview. Inflamm. Bowel Dis. 2005, 11, 927–936.
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550.
- Vaiopoulou, A.; Gazouli, M.; Theodoropoulos, G.; Zografos, G. Current advantages in the application of proteomics in inflammatory bowel disease. Dig. Dis. Sci. 2012, 57, 2755–2764.
- Gisbert, J.P.; Chaparro, M. Clinical Usefulness of Proteomics in Inflammatory Bowel Disease: A Comprehensive Review. J. Crohns Colitis 2019, 13, 374–384.
- Chan, P.P.; Wasinger, V.C.; Leong, R.W. Current application of proteomics in biomarker discovery for inflammatory bowel disease. World J. Gastrointest. Pathophysiol. 2016, 7, 27–37.
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207.
- Zhang, H.; Ge, Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ. Cardiovasc. Genet. 2011, 4, 711.
- Naryzhny, S. Inventory of proteoforms as a current challenge of proteomics: Some technical aspects. J. Proteom. 2019, 191, 22–28.
- Durbin, K.R.; Fornelli, L.; Fellers, R.T.; Doubleday, P.F.; Narita, M.; Kelleher, N.L. Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. J. Proteome Res. 2016, 15, 976–982.
- Baggerman, G.; Vierstraete, E.; De Loof, A.; Schoofs, L. Gel-based versus gel-free proteomics: A review. Comb. Chem. High Throughput Screen. 2005, 8, 669–677.
- Westbrook, J.A.; Noirel, J.; Brown, J.E.; Wright, P.C.; Evans, C.A. Quantitation with chemical tagging reagents in biomarker studies. Proteom. Clin. Appl. 2015, 9, 295–300.
- Canas, B.; Pineiro, C.; Calvo, E.; Lopez-Ferrer, D.; Gallardo, J.M. Trends in sample preparation for classical and second generation proteomics. J. Chromatogr. A 2007, 1153, 235–258.
- Alex, P.; Gucek, M.; Li, X. Applications of proteomics in the study of inflammatory bowel diseases: Current status and future directions with available technologies. Inflamm. Bowel Dis. 2009, 15, 616–629.
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196.
- Coskun, O. Separation techniques: Chromatography. North Clin. Istanb. 2016, 3, 156–160.
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126.
- Bateman, N.W.; Goulding, S.P.; Shulman, N.J.; Gadok, A.K.; Szumlinski, K.K.; MacCoss, M.J.; Wu, C.C. Maximizing peptide identification events in proteomic workflows using data-dependent acquisition (DDA). Mol. Cell. Proteom. 2014, 13, 329–338.
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169.
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904.
- Ong, S.E.; Mann, M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol. Biol. 2007, 359, 37–52.
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386.
- Schoeters, F.; Van Dijck, P. Protein-Protein Interactions in Candida albicans. Front. Microbiol. 2019, 10, 1792.
- Han, X.; Aslanian, A.; Yates, J.R., 3rd. Mass spectrometry for proteomics. Curr. Opin. Chem. Biol. 2008, 12, 483–490.
- Olshina, M.A.; Sharon, M. Mass Spectrometry: A Technique of Many Faces. Q. Rev. Biophys. 2016, 49, e18.
- Nadler, W.M.; Waidelich, D.; Kerner, A.; Hanke, S.; Berg, R.; Trumpp, A.; Rosli, C. MALDI versus ESI: The Impact of the Ion Source on Peptide Identification. J. Proteome Res. 2017, 16, 1207–1215.
More
Information
Subjects:
Gastroenterology & Hepatology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
603
Revisions:
2 times
(View History)
Update Date:
12 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No