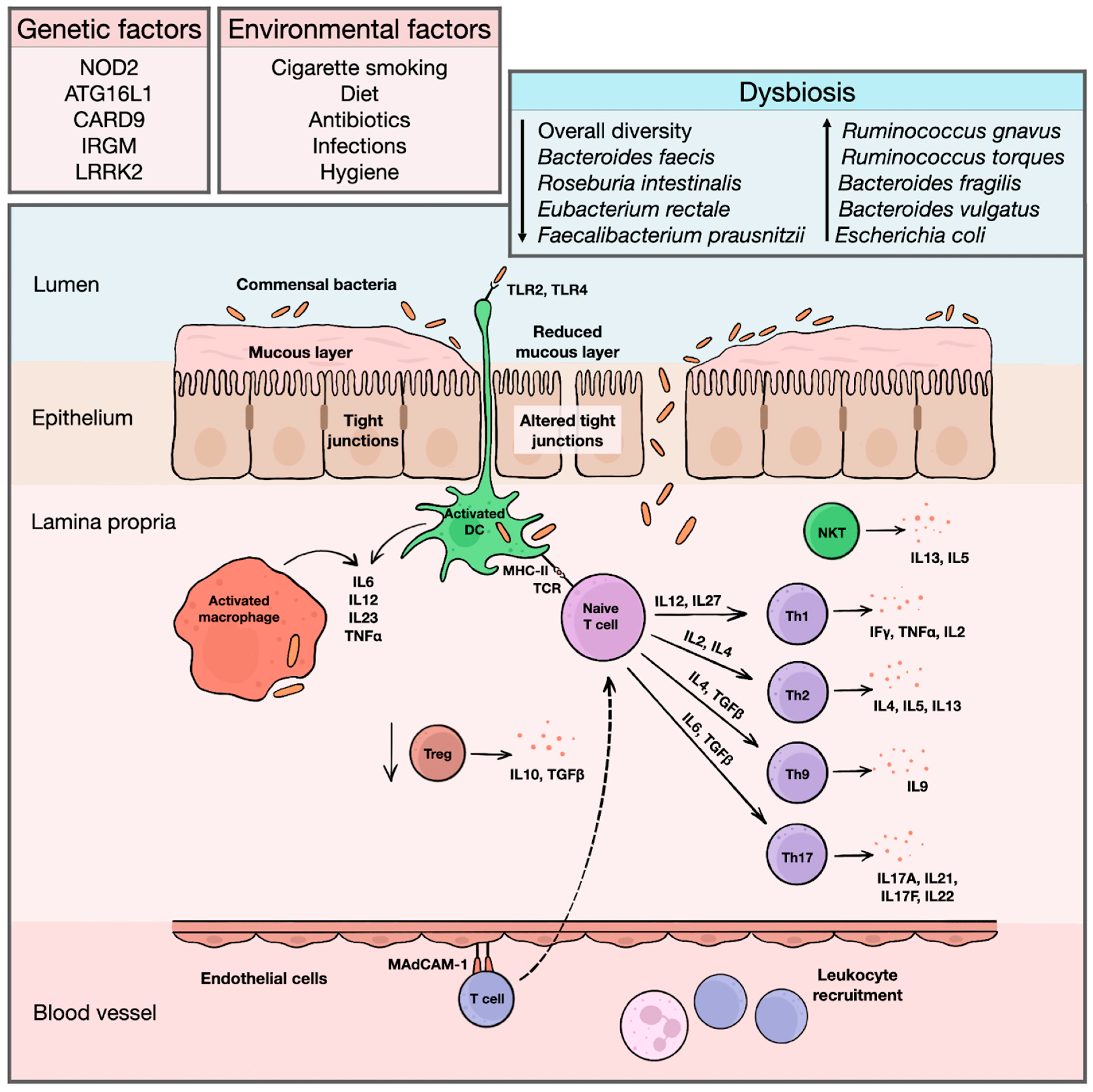
Inflammatory bowel diseases (IBD) are systemic immune-mediated conditions with predilection for the gastrointestinal tract and include Crohn’s disease and ulcerative colitis. Genomic and transcriptomic studies contributed substantially to our understanding of the immunopathological pathways involved in disease initiation and progression. However, eventual genomic alterations do not necessarily translate into the final clinical picture. Proteomics represent a missing link between the genome, transcriptome, and phenotypical presentation of the disease. Based on the analysis of a large spectrum of proteins in tissues, it seems to be a promising method for the identification of new biomarkers.
- inflammatory bowel disease
- pediatric
- proteomics
- proteome
- ulcerative colitis
1. Introduction
2. The Value of Proteomics
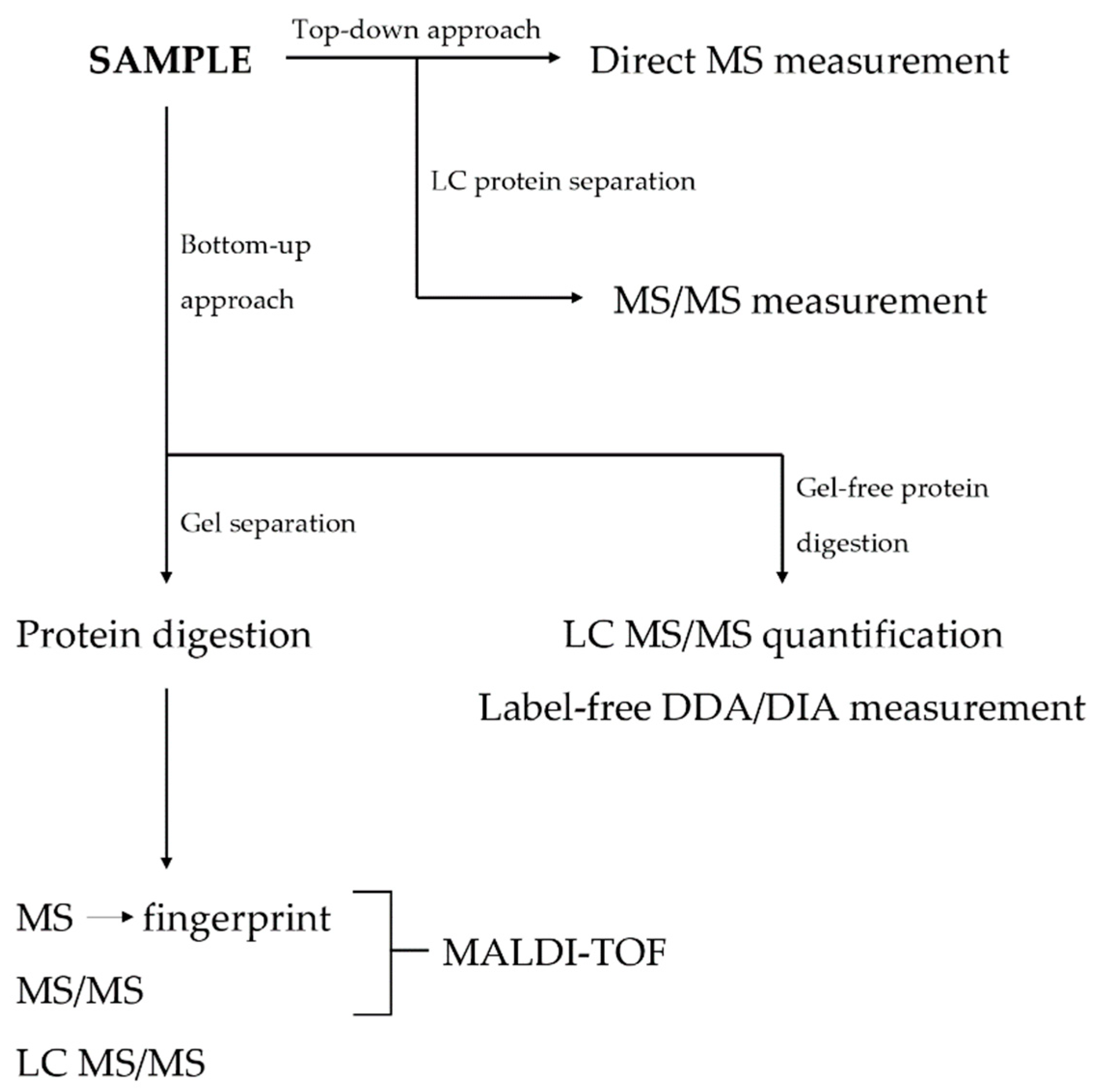
3. Techniques in Proteomics
This entry is adapted from the peer-reviewed paper 10.3390/ijms24119386
References
- Magro, F.; Langner, C.; Driessen, A.; Ensari, A.; Geboes, K.; Mantzaris, G.J.; Villanacci, V.; Becheanu, G.; Borralho Nunes, P.; Cathomas, G.; et al. European consensus on the histopathology of inflammatory bowel disease. J. Crohns Colitis 2013, 7, 827–851.
- Levine, A.; Koletzko, S.; Turner, D.; Escher, J.C.; Cucchiara, S.; de Ridder, L.; Kolho, K.L.; Veres, G.; Russell, R.K.; Paerregaard, A.; et al. ESPGHAN revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 795–806.
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54.e42, quiz e30.
- Benchimol, E.I.; Fortinsky, K.J.; Gozdyra, P.; Van den Heuvel, M.; Van Limbergen, J.; Griffiths, A.M. Epidemiology of pediatric inflammatory bowel disease: A systematic review of international trends. Inflamm. Bowel Dis. 2011, 17, 423–439.
- Burisch, J.; Pedersen, N.; Cukovic-Cavka, S.; Brinar, M.; Kaimakliotis, I.; Duricova, D.; Shonova, O.; Vind, I.; Avnstrom, S.; Thorsgaard, N.; et al. East-West gradient in the incidence of inflammatory bowel disease in Europe: The ECCO-EpiCom inception cohort. Gut 2014, 63, 588–597.
- Conrad, M.A.; Rosh, J.R. Pediatric Inflammatory Bowel Disease. Pediatr. Clin. North Am. 2017, 64, 577–591.
- Prenzel, F.; Uhlig, H.H. Frequency of indeterminate colitis in children and adults with IBD—A metaanalysis. J. Crohns Colitis 2009, 3, 277–281.
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640.
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983.
- Tyers, M.; Mann, M. From genomics to proteomics. Nature 2003, 422, 193–197.
- Assadsangabi, A.; Evans, C.A.; Corfe, B.M.; Lobo, A. Application of Proteomics to Inflammatory Bowel Disease Research: Current Status and Future Perspectives. Gastroenterol. Res. Pract. 2019, 2019, 1426954.
- Baldan-Martin, M.; Chaparro, M.; Gisbert, J.P. Tissue Proteomic Approaches to Understand the Pathogenesis of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1184–1200.
- Bennike, T.; Birkelund, S.; Stensballe, A.; Andersen, V. Biomarkers in inflammatory bowel diseases: Current status and proteomics identification strategies. World J. Gastroenterol. 2014, 20, 3231–3244.
- Lawrance, I.C.; Klopcic, B.; Wasinger, V.C. Proteomics: An overview. Inflamm. Bowel Dis. 2005, 11, 927–936.
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550.
- Vaiopoulou, A.; Gazouli, M.; Theodoropoulos, G.; Zografos, G. Current advantages in the application of proteomics in inflammatory bowel disease. Dig. Dis. Sci. 2012, 57, 2755–2764.
- Gisbert, J.P.; Chaparro, M. Clinical Usefulness of Proteomics in Inflammatory Bowel Disease: A Comprehensive Review. J. Crohns Colitis 2019, 13, 374–384.
- Chan, P.P.; Wasinger, V.C.; Leong, R.W. Current application of proteomics in biomarker discovery for inflammatory bowel disease. World J. Gastrointest. Pathophysiol. 2016, 7, 27–37.
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207.
- Zhang, H.; Ge, Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ. Cardiovasc. Genet. 2011, 4, 711.
- Naryzhny, S. Inventory of proteoforms as a current challenge of proteomics: Some technical aspects. J. Proteom. 2019, 191, 22–28.
- Durbin, K.R.; Fornelli, L.; Fellers, R.T.; Doubleday, P.F.; Narita, M.; Kelleher, N.L. Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. J. Proteome Res. 2016, 15, 976–982.
- Baggerman, G.; Vierstraete, E.; De Loof, A.; Schoofs, L. Gel-based versus gel-free proteomics: A review. Comb. Chem. High Throughput Screen. 2005, 8, 669–677.
- Westbrook, J.A.; Noirel, J.; Brown, J.E.; Wright, P.C.; Evans, C.A. Quantitation with chemical tagging reagents in biomarker studies. Proteom. Clin. Appl. 2015, 9, 295–300.
- Canas, B.; Pineiro, C.; Calvo, E.; Lopez-Ferrer, D.; Gallardo, J.M. Trends in sample preparation for classical and second generation proteomics. J. Chromatogr. A 2007, 1153, 235–258.
- Alex, P.; Gucek, M.; Li, X. Applications of proteomics in the study of inflammatory bowel diseases: Current status and future directions with available technologies. Inflamm. Bowel Dis. 2009, 15, 616–629.
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196.
- Coskun, O. Separation techniques: Chromatography. North Clin. Istanb. 2016, 3, 156–160.
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126.
- Bateman, N.W.; Goulding, S.P.; Shulman, N.J.; Gadok, A.K.; Szumlinski, K.K.; MacCoss, M.J.; Wu, C.C. Maximizing peptide identification events in proteomic workflows using data-dependent acquisition (DDA). Mol. Cell. Proteom. 2014, 13, 329–338.
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169.
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904.
- Ong, S.E.; Mann, M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol. Biol. 2007, 359, 37–52.
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386.
- Schoeters, F.; Van Dijck, P. Protein-Protein Interactions in Candida albicans. Front. Microbiol. 2019, 10, 1792.
- Han, X.; Aslanian, A.; Yates, J.R., 3rd. Mass spectrometry for proteomics. Curr. Opin. Chem. Biol. 2008, 12, 483–490.
- Olshina, M.A.; Sharon, M. Mass Spectrometry: A Technique of Many Faces. Q. Rev. Biophys. 2016, 49, e18.
- Nadler, W.M.; Waidelich, D.; Kerner, A.; Hanke, S.; Berg, R.; Trumpp, A.; Rosli, C. MALDI versus ESI: The Impact of the Ion Source on Peptide Identification. J. Proteome Res. 2017, 16, 1207–1215.