Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Suma Sarojini | -- | 2296 | 2023-06-08 11:19:50 | | | |
| 2 | Dean Liu | Meta information modification | 2296 | 2023-06-09 02:14:26 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Biswas, S.; Sarojini, S.; Jayaram, S.; Philip, I.; Umesh, M.; Mascarenhas, R.; Pappuswamy, M.; Balasubramanian, B.; Arokiyaraj, S. Role of Antimicrobial Peptides in Autoimmune Disorders. Encyclopedia. Available online: https://encyclopedia.pub/entry/45329 (accessed on 26 June 2026).
Biswas S, Sarojini S, Jayaram S, Philip I, Umesh M, Mascarenhas R, et al. Role of Antimicrobial Peptides in Autoimmune Disorders. Encyclopedia. Available at: https://encyclopedia.pub/entry/45329. Accessed June 26, 2026.
Biswas, Soma, Suma Sarojini, Saranya Jayaram, Indhu Philip, Mridul Umesh, Roseanne Mascarenhas, Manikantan Pappuswamy, Balamuralikrishnan Balasubramanian, Selvaraj Arokiyaraj. "Role of Antimicrobial Peptides in Autoimmune Disorders" Encyclopedia, https://encyclopedia.pub/entry/45329 (accessed June 26, 2026).
Biswas, S., Sarojini, S., Jayaram, S., Philip, I., Umesh, M., Mascarenhas, R., Pappuswamy, M., Balasubramanian, B., & Arokiyaraj, S. (2023, June 08). Role of Antimicrobial Peptides in Autoimmune Disorders. In Encyclopedia. https://encyclopedia.pub/entry/45329
Biswas, Soma, et al. "Role of Antimicrobial Peptides in Autoimmune Disorders." Encyclopedia. Web. 08 June, 2023.
Copy Citation
The antimicrobial peptides are present in many parts of the human body such as skin, mucosae, etc., that are exposed to microbes. AMPs are typically produced together as a mixture of several peptides, with tissue-specific unique AMP combinations. While specific AMPs are more prevalent in particular parts of the body, very few are exclusively produced by a single tissue or cell type. Almost all AMPs have multiple functions. AMPs, such as defensins and cathelicidins, were initially identified and studied due to their antimicrobial properties. Defensins and cathelicidins possess various immunomodulatory activities apart from their broad spectrum of activity against pathogens.
antimicrobial peptides
neutrophil
autoimmune disorders
defensins
cathelicidins
1. Rheumatic Arthritis (RA)
RA is a type of inflammatory condition that affects joints and causes long-term damage to cartilage and bones. It is caused due to the accumulation of autoantibodies. In RA patients, α-defensins 1–3 (HNPs) were identified in synovial fluid, which acts as biomarkers [1]. In rats with arthritis, the synovial membranes in their joints had the highest concentration of cathelicidins. Neutrophils, a type of immune cell, are pushed into the inflamed joints by granulocyte colony-stimulating factor, which also triggers their production. Leukotriene B4 and its receptor promote the recruitment of neutrophils into the joints by causing dysregulation of T-helper type 17 [2]. Neutrophil activation leads to the production of cytokine IL-1β in synovial fluid which then produces chemokines and promotes neutrophil recruitment into the joints. Rescinding of RA may be possible by reducing neutrophils moving towards the synovial fluid by making changes in neutrophil adhesion and chemotaxis in the joints. Neutrophils in the bloodstream and synovial fluid are more likely to form NETs [3]. RA NETs promote inflammation by activating cytokines, chemokines, and adhesion molecules in synovial fluids. In RA, the level of NETosis is related to anti-citrullinated peptide antibodies [4]. The proinflammatory cytokines such as TNF-α, IL-6, and IL-1β can activate the production of HBD-2 in chondrocytes. HBD3, another β-defensin, activates metalloproteinases by chondrocytes that play an important role in cartilage destruction (Figure 1). Elevated levels of the human cathelicidin LL-37 and its processing enzyme proteinase 3 were observed to be associated with this disease. LL-37 also triggers apoptosis of osteoblasts, which results in reduced bone formation in the joints affected by arthritis [1]. The pathogenesis of rheumatoid arthritis gets triggered by neutrophil-releasing substances that promote inflammation and attract immune cells. The complete etiology and pathogenesis of RA have not been completely elucidated, but the use of some anti-rheumatic medications such as adalimumab and etanercept has been shown to noticeably decrease the expression of LL-37. This suggests a connection between LL-37 expression and the severity of RA [5].
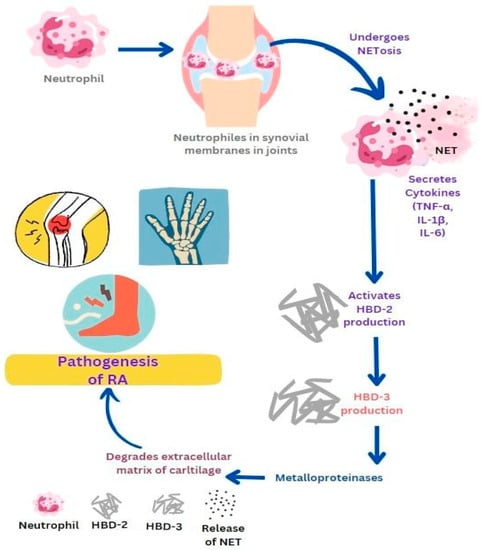
Figure 1. Role of antimicrobial peptides in promoting the pathogenesis of rheumatoid arthritis (RA). Neutrophil activation in joints leads to the production of cytokines and chemokines which further promote neutrophil recruitment and NETs formation. Activated NETs promote inflammation by producing cytokines such as TNF-α, IL-6, and IL-1β which then activate the production of HBD-2 and HBD-3. HBD-3 activates metalloproteinases which play an important role in cartilage destruction.
2. Systemic Lupus Erythematosus (SLE)
SLE is a multifactorial autoimmune problem that affects the entire body and occurs when the immune system loses control and attacks the body’s own tissues. It is caused by the presence of autoantibodies that target nuclear antigens. SLE is characterized by symptoms that potentially attack almost every organ system, although it primarily affects the heart, joints, skin, lungs, blood vessels, liver, kidneys, and nervous system. Increased NETosis by SLE neutrophils were observed upon stimulation by antibodies against LL-37 [6]. In SLE, neutrophils are thought to be responsible for the pathogenesis of the disease. Activated neutrophils release AMPs that complexed with self-DNA and trigger pDCs activation via TLR9. AMP-self-DNA complexes in NETs possibly act as auto-antigens against which auto-antibodies are made in SLE. A link has been proposed between the activation of neutrophils, pDCs activation, and the development of autoimmunity in SLE [3]. Activation of pDCs releases type I interferon-α which increases the monocytes’ differentiation into dendritic cells, which subsequently stimulate the activation of self-reactive B and T cells, thereby promoting autoimmunity in SLE. High numbers of apoptotic neutrophils, as well as immature neutrophils, were detected in patients’ blood. Self-DNA are very unstable in the extracellular environment and degrade easily [7], but when self-DNA are complexed in an immunogenic complex, they trigger the activation of pDCs via TLR9.
Possibly, AMPs such as LL-37 and HNPs play an important role in making self-DNA immunogenic by inducing aggregation of self-DNA into an insoluble particle, against which auto-antibodies are released. These immunogenic complexes having self-DNA and AMPs are released from apoptotic neutrophils during NETosis (Figure 2) [1]. In SLE patients, auto-antibodies are released not only against self-DNA but also against AMPs in NETs. These auto-antibodies in the immune complex interact with FcGamma RII on pDCs and trigger receptor-mediated endocytosis of self-DNA. Neutrophils in SLE patients express high amounts of surface LL-37 and HNPs against which antibodies are secreted. Anti-LL-37 and anti-HNP trigger the release of NETs from neutrophils. Thus, circulating neutrophils in SLE release large amounts of NETs in response to circulating antibodies against AMPs [8]. In patients with SLE, neutrophil activation results in α-defensin secretion. These, in turn, function as chemoattractants for T-lymphocytes, mononuclear cells, and dendritic cells. These together activate the innate and adaptive immunity leading to the diseased state [1]. The pathogenicity of SLE stems from the involvement of cathelicidin, its presence in NETs, and its capability to create and maintain immune complexes with DNA and autoantibodies. As mentioned earlier, these complexes stimulate the secretion of type I interferon (IFN) by plasmacytoid dendritic cells (pDCs) and the production of autoantibodies by B cells [9]. Nevertheless, the precise role of each cell in SLE is not yet completely understood.
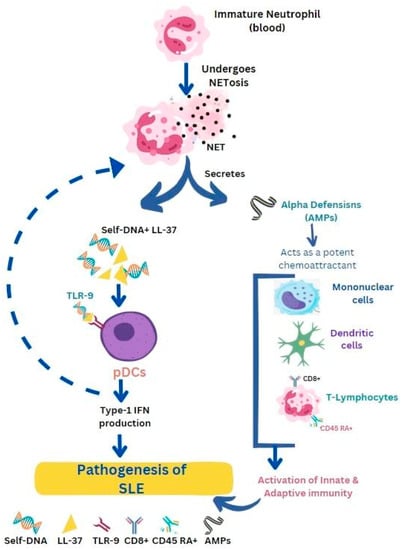
Figure 2. Antimicrobial peptides such as LL-37 and defensins facilitate the progression of systemic lupus erythematosus (SLE). Release of immunogenic complexes (self DNA + LL-37) from apoptotic neutrophils prompts the activation of pDCs via Toll-like receptors (TLR-9). Activated pDCs release type-1 IFN which further promotes NETosis. α-defensin secretion from activated neutrophils serves as a chemoattractant for immune cells, activating innate and adaptive immunity leading to the SLE condition.
3. Type I Diabetes Mellitus (TIDM)
TIDM is a disease caused by an autoimmune response that destroys the pancreatic β-cells responsible for producing insulin. In TIDM, the T cells that are self-reactive activate the insulin-producing β-cells in the pancreas. Levels of LL-37 and β-defensins (HBD1) levels are decreased in comparison to healthy individuals. Studies show that AMP dysregulation might lead to TIDM [1]. Research has also demonstrated the crucial role of gut microbiota in the pancreas in regulating autoimmune diabetes. Gut microbiota helps to modulate CRAMP production; CRAMP regulates the pancreatic immune environment by inducing lymphoid and myeloid regulatory cells to prevent autoimmune diabetes [10][11].
Mouse studies have elucidated the involvement of gut microbiota in the development of autoimmune TIDM. Reduction of CRAMP expression caused dysbiosis and an increase in pancreatic autoimmune response, further initiating the development of diabetes. An increase in CRAMP expression by local treatment reinstated colonic homeostasis. Infections by enteric pathogens can upset gut barriers leading to the development of TIDM [12]. Citrobacter rodentium was used as a model organism to represent colonic infection with disruption in the gut barrier. The infection caused a reduction of CRAMP production, activation in the interferon-gamma (IFN-γ)+ cells, and an increase in T1DM. They concluded that cathelicidin supplementation can bring back healthy gut barriers and thus prevent T1DM [13]. Like cathelicidins, β defensins also show immunomodulatory properties such as chemotaxis, immune cell modulation, and regulation of both adaptive and innate immune responses. The gut microbiota has the ability to regulate the expression of CRAMP in pancreatic β-cells by producing short-chain fatty acids. Therefore, modifying the gut microbiota could be a potential approach to reduce the pathogenesis of autoimmune diabetes by altering the expression of AMPs [12]. In a study, Miani et al. [14] reported the role of mBD14 (mouse β-defensin 14) against T1DM. The AMPs stimulated the production of IL-4 by B-cells and caused the β cells of the pancreas to express transgenic IL-4. As a result, diabetes was prevented in NOD (non-obese diabetics) mice [14]. Modifying intestinal antimicrobial peptides could be seen as a significant therapeutic strategy to safeguard at-risk children from developing autoimmune diabetes.
4. Psoriasis
Psoriasis is a chronic inflammatory disease with symptoms affecting the skin with the presence of inflammatory plaques and characterized by complex immune responses wherein molecular, cellular, and environmental factors play important roles in the disease development. Injury, infection, or genetic factors trigger the keratinocytes to produce elevated concentrations of AMPs such as LL-37, which can bind to self-DNA to form an immunogenic complex that activates pDCs through TLR9. Increased TLR9 production and expression leads to increased type I IFN and TNF-α expression and inflammation. Human cathelicidin (LL-37) can form a complex with self-RNA to activate pDCs through TLR7 and mDCs through TLR8. Additionally, neutrophils are also capable of inducing the expression of human β-defensin 2 (HBD2) in keratinocytes of psoriatic skin through the formation of NETs [15][16]. LL-37 is also identified as a self-antigen for circulating T-cells in psoriasis.
Circulating auto-antibodies were also found in psoriasis patients. Human keratinocytes have Mas-related G-protein receptors. These receptors get continuously activated by LL-37 and β-defensins leading to the generation of cytokines and inflammation [1]. Studies show that PMNs respond quickly to RNA-LL-37 as compared to DNA-LL-37. Upon encountering RNA-LL-37 cytokines, chemokines, and NETs are released via TLR8 and TLR13. The NETs released by PMN also contain a high amount of RNA. Thus, PMNs and NET-derived RNA-LL-37 act as contributors to the self-propagating inflammatory cycle in psoriasis [17]. In psoriasis, LL-37 interacts with both self-DNA and self-RNA and translocates them to endosomal compartments of dendritic cells. Self-RNA-LL-37 triggers the activation of pDCs through TLR7 with the production of IFN-α, while self-DNA-LL-37 activates TLR9 with the release of IFN-α from pDCs. Self-RNA-LL-37 also activates TLR8 and in response releases inflammatory cytokines IL-6, IL-12, and IL-23 from mDCs (Figure 3). LL-37 acts as a stimulator to convert non-stimulatory self-RNA released from dying host cells to take part in innate immune activation. LL-37, when interacting with self-RNA forms, aggregates that protect the RNA against extracellular digestion by RNase [9][18]. In psoriasis, IL-17 is produced by innate immune cells and ETs found in human skin. Mast cells when treated with IL-23 and IL-1β in human skin readily form ETs with the release of IL-17. Mast cells and neutrophils have a significant role in psoriasis, as they release IL-17 through the formation of NETs [19] LL-37 which acts as an autoantigen in psoriasis and has the ability to directly initiate T cell activation. Apart from neutrophils and plasmacytoid dendritic cells (pDCs), other innate lymphocytes such as natural killer T (NKT) cells and natural killer (NK) cells that produce AMPs also have crucial roles in the development of psoriasis [5]. Human keratinocytes exhibit high levels of expression for Mas-related G protein-coupled receptors MRGPRX3 and MRGPRX4. The uncontrolled activation of these receptors by human defense peptides such as β-defensins and LL-37 can contribute to sensations of itch, pain, and other chronic inflammatory skin diseases [1]. In summary, AMPs and the cells that produce them play a significant part in the pathogenesis of psoriasis.
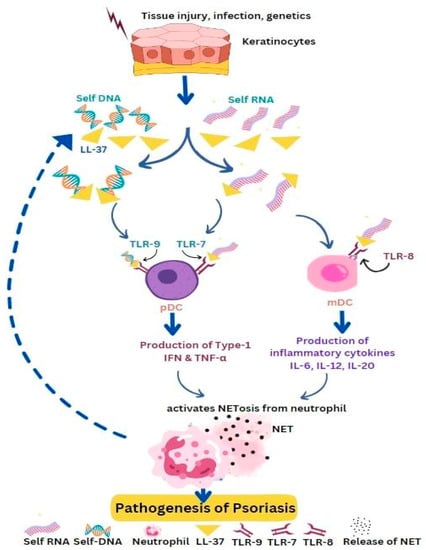
Figure 3. Self-DNA/LL-37 and self-RNA/LL-37 promote the pathogenesis of psoriasis. Keratinocytes get activated under the pressure of tissue injury, infection, or genetic factors and express high levels of AMPs and nucleic acids, which form immunogenic complexes that activate pDCs or mDCs via Toll-like receptors (TLR9 or TLR8). Activated DCs produce type I IFN, TNF-α, and cytokines (IL-6, IL-12, IL-23) which promote NETosis. Activated neutrophils produce LL-37 and nucleic acids, thereby self-propagating the inflammatory cycle.
5. Vasculitis
Anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis (AAV) refers to a group of autoimmune diseases that affect the entire body and are characterized by inflammation of small- to medium-sized blood vessels, causing necrotizing vasculitis [20]. Myeloperoxidase (MPO) and proteinase-3 (PR3) are proteins found in the lysosomes of monocytes and primary granules of neutrophils. When ANCA activates these cells, MPO and PR3 are released [21]. Zhang and colleagues were the first to discover that cathelicidin LL37 levels are elevated in AAV, particularly in patients with crescentic glomerulonephritis [22]. Cathelicidin LL37 is stored in an inactive form in the secondary granules of neutrophils and activated by PR3 (the target of PR3-ANCA) when released during inflammation [23]. When neutrophils and monocytes are activated by ANCA, they release MPO and PR3 along with other lysosome and granule proteins [21]. In addition, activated neutrophils also release cathelicidin LL37, which is found in neutrophil extracellular traps that have been linked to autoimmunity induction and inflammation mediation in AAV [24]. Therefore, cathelicidin LL37 can serve as valuable indicators in detecting active AAV, such as aggressive crescentic glomerulonephritis, making them possible prognostic markers to develop novel therapies [22].
6. Gout
Gout is a type of inflammatory arthritis accompanied by painful joints. It is caused when the body’s own inflammatory response is activated by the accumulation of monosodium urate (MSU) crystals in the joints. These crystals attract immune cells, known as leukocytes, and stimulate the production of NETs, which in turn lead to inflammation [25][26]. Auto-inflammatory disease such as gout has been associated with NETosis and aggregation of NETs. It is proposed that the clustering of NETs aids in the subsiding of inflammation caused by neutrophils by breaking down cytokines and chemokines and interfering with the recruitment and activation of neutrophils [27]. The presence of MSU crystals triggers an inflammatory response by activating the NALP3 inflammasome in macrophages that reside in the tissue. This, in turn, leads to the production of a significant amount of IL-1β. Consistent with previous research, the components NLRP3, ASC, Caspase-1, IL-1β, and IL-1R are all crucial factors in the inflammation and hypersensitivity that results from MSU crystal-induced joint inflammation [28][29]. The role of cathelicidin LL-37 on the activation of NLRP3 inflammasome is still not clear [30]. The complete understanding of the association between LL-37 and clinical markers, as well as pro-inflammatory mediators in individuals diagnosed with gout, remains uncertain.
References
- Umnyakova, E.S.; Zharkova, M.S.; Berlov, M.N.; Shamova, O.V.; Kokryakov, V.N. Human antimicrobial peptides in autoimmunity. Autoimmunity 2020, 53, 137–147.
- Hoffmann, M.H.; Bruns, H.; Bäckdahl, L.; Neregård, P.; Niederreiter, B.; Herrmann, M.; Catrina, A.I.; Agerberth, B.; Holmdahl, R. The cathelicidins LL-37 and rCRAMP are associated with pathogenic events of arthritis in humans and rats. Ann. Rheum. Dis. 2013, 72, 1239–1248.
- Kaplan, M.J. Role of neutrophils in systemic autoimmune diseases. Arthritis Res. Ther. 2013, 15, 219.
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131.
- Zhang, C.; Yang, M. The Role and Potential Application of Antimicrobial Peptides in Autoimmune Diseases. Front. Immunol. 2020, 11, 859.
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81.
- Apel, F.; Zychlinsky, A.; Kenny, E.F. The role of neutrophil extracellular traps in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 467–475.
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569.
- Liang, W.; Diana, J. The Dual Role of Antimicrobial Peptides in Autoimmunity. Front. Immunol. 2020, 11, 2077.
- Sun, J.; Furio, L.; Mecheri, R.; van der Does, A.M.; Lundeberg, E.; Saveanu, L.; Chen, Y.; van Endert, P.; Agerberth, B.; Diana, J. Pancreatic β-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed under the Influence of the Gut Microbiota. Immunity 2015, 43, 304–317.
- Depta, J.; Małkowska, P.; Wysokinska, M.; Todorska, K.; Sierawska, O.; Hrynkiewicz, R.; Bebnowska, D.; Niedzwiedzka-Rystwej, P. Therapeutic Role of Antimicrobial Peptides in Diabetes Mellitus. Biologics 2022, 2, 92–106.
- Liang, W.; Enée, E.; Andre-Vallee, C.; Falcone, M.; Sun, J.; Diana, J. Intestinal cathelicidin antimicrobial peptide shapes a protective neonatal gut microbiota against pancreatic autoimmunity. Gastroenterology 2022, 162, 1288–1302.
- Jia, L.; Jiahong, L.; Ming, Z.; He, L.; Zhengnan, R.; Xiao, L.; Xiaohua, P.; Ju, Q.; Li-Long, P.; Jia, S. Cathelicidin-related antimicrobial peptide protects against enteric pathogen-accelerated type 1 diabetes in mice. Theranostics 2022, 12, 3438.
- Miani, M.; Julie, L.; Emmanuelle, W.; Subash, V.; Marjolène, S.; Patrick, E.; Bernhard, R.; Peter, E.; Harry, S.; Julien, D. Gut microbiota-stimulated innate lymphoid cells support β-defensin 14 expression in pancreatic endocrine cells, preventing autoimmune diabetes. Cell. Metab. 2018, 28, 557–572.
- Eisenbeis, J.; Saffarzadeh, M.; Peisker, H.; Jung, P.; Thewes, N.; Preissner, K.T.; Herrmann, M.; Molle, V.; Geisbrecht, B.V.; Jacobs, K.; et al. The Staphylococcus aureus Extracellular Adherence Protein Eap Is a DNA Binding Protein Capable of Blocking Neutrophil Extracellular Trap Formation. Front. Cell. Infect. Microbiol. 2018, 8, 235.
- Burgener, S.S.; Schroder, K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb. Perspect. Biol. 2020, 12, a037028.
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhöfer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Löffler, M.W.; Kalbacher, H.; et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 105.
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994.
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 2011, 187, 490–500.
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013, 65, 1–11.
- Jennette, J.C.; Falk, R.J.; Gasim, A.H. Pathogenesis of antineutrophil cytoplasmic autoantibody vasculitis. Curr. Opin. Nephrol. Hypertens. 2011, 20, 263–270.
- Zhang, Y.; Shi, W.; Tang, S.; Li, J.; Yin, S.; Gao, X.; Wang, L.; Zou, L.; Zhao, J.; Huang, Y.; et al. The influence of cathelicidin LL37 in human anti-neutrophils cytoplasmic antibody (ANCA)-associated vasculitis. Arthritis Res. Ther. 2013, 15, R161.
- Jann, N.J.; Schmaler, M.; Kristian, S.A.; Radek, K.A.; Gallo, R.L.; Nizet, V.; Peschel, A.; Landmann, R. Neutrophil antimicrobial defense against Staphylococcus aureus is mediated by phagolysosomal but not extracellular trapassociated cathelicidin. J. Leukoc. Biol. 2009, 86, 1159–1169.
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625.
- Desai, J.; Steiger, S.; Anders, H.J. Molecular Pathophysiology of Gout. Trends Mol. Med. 2017, 23, 756–768.
- Hidalgo, A.I.; Carretta, M.D.; Alarcón, P.; Manosalva, C.; Müller, A.; Navarro, M.; Hidalgo, M.A.; Kaehne, T.; Taubert, A.; Hermosilla, C.R.; et al. Proinflammatory mediators and neutrophils are increased in synovial fluid from heifers with acute ruminal acidosis. BMC Vet. Res. 2019, 15, 225.
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517.
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241.
- Amaral, F.A.; Costa, V.V.; Tavares, L.D.; Sachs, D.; Coelho, F.M.; Fagundes, C.T.; Soriani, F.M.; Silveira, T.N.; Cunha, L.D.; Zamboni, D.S.; et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout. Arthritis Rheum. 2012, 64, 474–484.
- Hu, Z.; Murakami, T.; Suzuki, K.; Tamura, H.; Reich, J.; Kuwahara-Arai, K.; Iba, T.; Nagaoka, I. Antimicrobial cathelicidin peptide LL-37 inhibits the pyroptosis of macrophages and improves the survival of polybacterial septic mice. Int. Immunol. 2016, 28, 245–253.
More
Information
Subjects:
Immunology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
772
Revisions:
2 times
(View History)
Update Date:
09 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No