The antimicrobial peptides are present in many parts of the human body such as skin, mucosae, etc., that are exposed to microbes. AMPs are typically produced together as a mixture of several peptides, with tissue-specific unique AMP combinations. While specific AMPs are more prevalent in particular parts of the body, very few are exclusively produced by a single tissue or cell type. Almost all AMPs have multiple functions. AMPs, such as defensins and cathelicidins, were initially identified and studied due to their antimicrobial properties. Defensins and cathelicidins possess various immunomodulatory activities apart from their broad spectrum of activity against pathogens.
- antimicrobial peptides
- neutrophil
- autoimmune disorders
- defensins
- cathelicidins
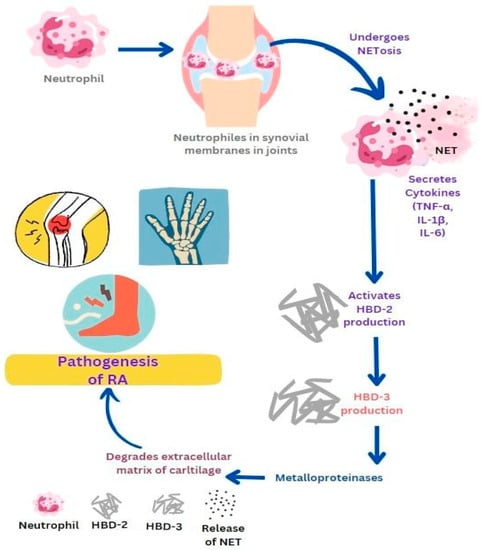
1. Rheumatic Arthritis (RA)
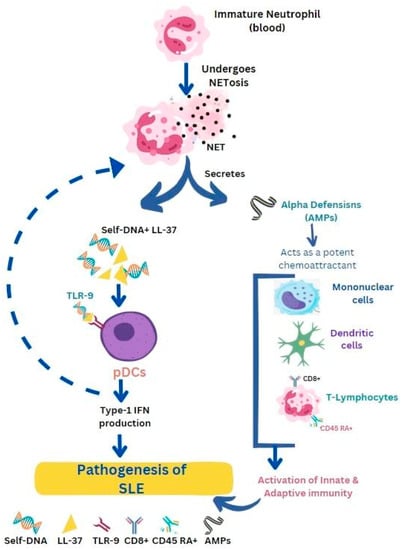
2. Systemic Lupus Erythematosus (SLE)
3. Type I Diabetes Mellitus (TIDM)
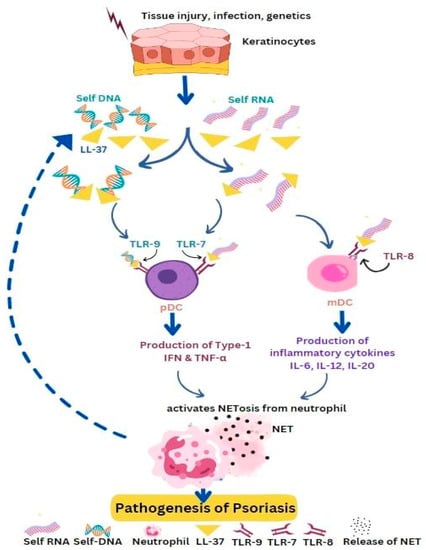
4. Psoriasis
5. Vasculitis
6. Gout
This entry is adapted from the peer-reviewed paper 10.3390/life13061307
References
- Umnyakova, E.S.; Zharkova, M.S.; Berlov, M.N.; Shamova, O.V.; Kokryakov, V.N. Human antimicrobial peptides in autoimmunity. Autoimmunity 2020, 53, 137–147.
- Hoffmann, M.H.; Bruns, H.; Bäckdahl, L.; Neregård, P.; Niederreiter, B.; Herrmann, M.; Catrina, A.I.; Agerberth, B.; Holmdahl, R. The cathelicidins LL-37 and rCRAMP are associated with pathogenic events of arthritis in humans and rats. Ann. Rheum. Dis. 2013, 72, 1239–1248.
- Kaplan, M.J. Role of neutrophils in systemic autoimmune diseases. Arthritis Res. Ther. 2013, 15, 219.
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131.
- Zhang, C.; Yang, M. The Role and Potential Application of Antimicrobial Peptides in Autoimmune Diseases. Front. Immunol. 2020, 11, 859.
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81.
- Apel, F.; Zychlinsky, A.; Kenny, E.F. The role of neutrophil extracellular traps in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 467–475.
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569.
- Liang, W.; Diana, J. The Dual Role of Antimicrobial Peptides in Autoimmunity. Front. Immunol. 2020, 11, 2077.
- Sun, J.; Furio, L.; Mecheri, R.; van der Does, A.M.; Lundeberg, E.; Saveanu, L.; Chen, Y.; van Endert, P.; Agerberth, B.; Diana, J. Pancreatic β-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed under the Influence of the Gut Microbiota. Immunity 2015, 43, 304–317.
- Depta, J.; Małkowska, P.; Wysokinska, M.; Todorska, K.; Sierawska, O.; Hrynkiewicz, R.; Bebnowska, D.; Niedzwiedzka-Rystwej, P. Therapeutic Role of Antimicrobial Peptides in Diabetes Mellitus. Biologics 2022, 2, 92–106.
- Liang, W.; Enée, E.; Andre-Vallee, C.; Falcone, M.; Sun, J.; Diana, J. Intestinal cathelicidin antimicrobial peptide shapes a protective neonatal gut microbiota against pancreatic autoimmunity. Gastroenterology 2022, 162, 1288–1302.
- Jia, L.; Jiahong, L.; Ming, Z.; He, L.; Zhengnan, R.; Xiao, L.; Xiaohua, P.; Ju, Q.; Li-Long, P.; Jia, S. Cathelicidin-related antimicrobial peptide protects against enteric pathogen-accelerated type 1 diabetes in mice. Theranostics 2022, 12, 3438.
- Miani, M.; Julie, L.; Emmanuelle, W.; Subash, V.; Marjolène, S.; Patrick, E.; Bernhard, R.; Peter, E.; Harry, S.; Julien, D. Gut microbiota-stimulated innate lymphoid cells support β-defensin 14 expression in pancreatic endocrine cells, preventing autoimmune diabetes. Cell. Metab. 2018, 28, 557–572.
- Eisenbeis, J.; Saffarzadeh, M.; Peisker, H.; Jung, P.; Thewes, N.; Preissner, K.T.; Herrmann, M.; Molle, V.; Geisbrecht, B.V.; Jacobs, K.; et al. The Staphylococcus aureus Extracellular Adherence Protein Eap Is a DNA Binding Protein Capable of Blocking Neutrophil Extracellular Trap Formation. Front. Cell. Infect. Microbiol. 2018, 8, 235.
- Burgener, S.S.; Schroder, K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb. Perspect. Biol. 2020, 12, a037028.
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhöfer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Löffler, M.W.; Kalbacher, H.; et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 105.
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994.
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J. Immunol. 2011, 187, 490–500.
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013, 65, 1–11.
- Jennette, J.C.; Falk, R.J.; Gasim, A.H. Pathogenesis of antineutrophil cytoplasmic autoantibody vasculitis. Curr. Opin. Nephrol. Hypertens. 2011, 20, 263–270.
- Zhang, Y.; Shi, W.; Tang, S.; Li, J.; Yin, S.; Gao, X.; Wang, L.; Zou, L.; Zhao, J.; Huang, Y.; et al. The influence of cathelicidin LL37 in human anti-neutrophils cytoplasmic antibody (ANCA)-associated vasculitis. Arthritis Res. Ther. 2013, 15, R161.
- Jann, N.J.; Schmaler, M.; Kristian, S.A.; Radek, K.A.; Gallo, R.L.; Nizet, V.; Peschel, A.; Landmann, R. Neutrophil antimicrobial defense against Staphylococcus aureus is mediated by phagolysosomal but not extracellular trapassociated cathelicidin. J. Leukoc. Biol. 2009, 86, 1159–1169.
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625.
- Desai, J.; Steiger, S.; Anders, H.J. Molecular Pathophysiology of Gout. Trends Mol. Med. 2017, 23, 756–768.
- Hidalgo, A.I.; Carretta, M.D.; Alarcón, P.; Manosalva, C.; Müller, A.; Navarro, M.; Hidalgo, M.A.; Kaehne, T.; Taubert, A.; Hermosilla, C.R.; et al. Proinflammatory mediators and neutrophils are increased in synovial fluid from heifers with acute ruminal acidosis. BMC Vet. Res. 2019, 15, 225.
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhöfer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517.
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241.
- Amaral, F.A.; Costa, V.V.; Tavares, L.D.; Sachs, D.; Coelho, F.M.; Fagundes, C.T.; Soriani, F.M.; Silveira, T.N.; Cunha, L.D.; Zamboni, D.S.; et al. NLRP3 inflammasome-mediated neutrophil recruitment and hypernociception depend on leukotriene B(4) in a murine model of gout. Arthritis Rheum. 2012, 64, 474–484.
- Hu, Z.; Murakami, T.; Suzuki, K.; Tamura, H.; Reich, J.; Kuwahara-Arai, K.; Iba, T.; Nagaoka, I. Antimicrobial cathelicidin peptide LL-37 inhibits the pyroptosis of macrophages and improves the survival of polybacterial septic mice. Int. Immunol. 2016, 28, 245–253.