Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Farid Khallouki | -- | 3945 | 2023-06-07 11:16:40 | | | |
| 2 | Lindsay Dong | Meta information modification | 3945 | 2023-06-08 05:35:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Khallouki, F.; Hajji, L.; Saber, S.; Bouddine, T.; Edderkaoui, M.; Bourhia, M.; Mir, N.; Lim, A.; El Midaoui, A.; Giesy, J.P.; et al. Role of Tamoxifen in Breast Cancer Management. Encyclopedia. Available online: https://encyclopedia.pub/entry/45283 (accessed on 27 June 2026).
Khallouki F, Hajji L, Saber S, Bouddine T, Edderkaoui M, Bourhia M, et al. Role of Tamoxifen in Breast Cancer Management. Encyclopedia. Available at: https://encyclopedia.pub/entry/45283. Accessed June 27, 2026.
Khallouki, Farid, Lhoussain Hajji, Somayya Saber, Toufik Bouddine, Mouad Edderkaoui, Mohammed Bourhia, Nora Mir, Adrian Lim, Adil El Midaoui, John P. Giesy, et al. "Role of Tamoxifen in Breast Cancer Management" Encyclopedia, https://encyclopedia.pub/entry/45283 (accessed June 27, 2026).
Khallouki, F., Hajji, L., Saber, S., Bouddine, T., Edderkaoui, M., Bourhia, M., Mir, N., Lim, A., El Midaoui, A., Giesy, J.P., Aboul-Soud, M.A.M., Silvente-Poirot, S., & Poirot, M. (2023, June 07). Role of Tamoxifen in Breast Cancer Management. In Encyclopedia. https://encyclopedia.pub/entry/45283
Khallouki, Farid, et al. "Role of Tamoxifen in Breast Cancer Management." Encyclopedia. Web. 07 June, 2023.
Copy Citation
Breast cancer (BC) is the most common female cancer in terms of incidence and mortality worldwide. Tamoxifen (Nolvadex) is a widely prescribed, oral anti-estrogen drug for the hormonal treatment of estrogen-receptor-positive BC, which represents 70% of all BC subtypes.
tamoxifen
estrogen receptor
breast cancer
1. Tamoxifen and Its Interaction with the ER
Estrogens, primarily produced in the ovarian follicles and adrenal glands, are the most important female sex hormones [1]. The estrogen receptor (ER) is a member of the nuclear receptor superfamily; its function, which is mediated by 17β-estradiol (E2), is to activate the transcription of genes involved in the growth and differentiation of cells, as well as reproduction.
The two distinct subtypes of ER, ERα and ERβ, each have different effects in gene regulation and cell proliferation. ERα is mainly present in tissues with reproductive functions, such as the ovaries, uterus, and breast, along with the brain, heart, liver, and bone. ERβ is detected in the ovarian, uterine, breast, hypothalamic, and cardiovascular tissues [2].
Structurally, ERs consist of six functional domains: A, B, C, D, E, and F, with three major domains including an N-terminal (A/B domain), a DNA-binding domain (C domain), and a ligand-binding domain (LBD, E domain) (Figure 1). The N-terminal (A/B domain) activation function (AF-1) region is involved in protein–protein interactions, is hormone-independent, and is mostly regulated by phosphorylation. The C-terminal (E domain) activation function (AF-2) contains the binding region for the ligand, along with co-activators or co-repressors, and is responsible for ligand-gated transcription. The highly conserved DNA-binding C domain consists of two zinc finger modules, which establishes contacts with specific response elements and is involved in the spatial recognition of the receptor. The flexible hinge or D domain contains a nuclear localization signal, acting as a dimerization region. Lastly, the F domain is variable, and its function is still poorly understood [1].
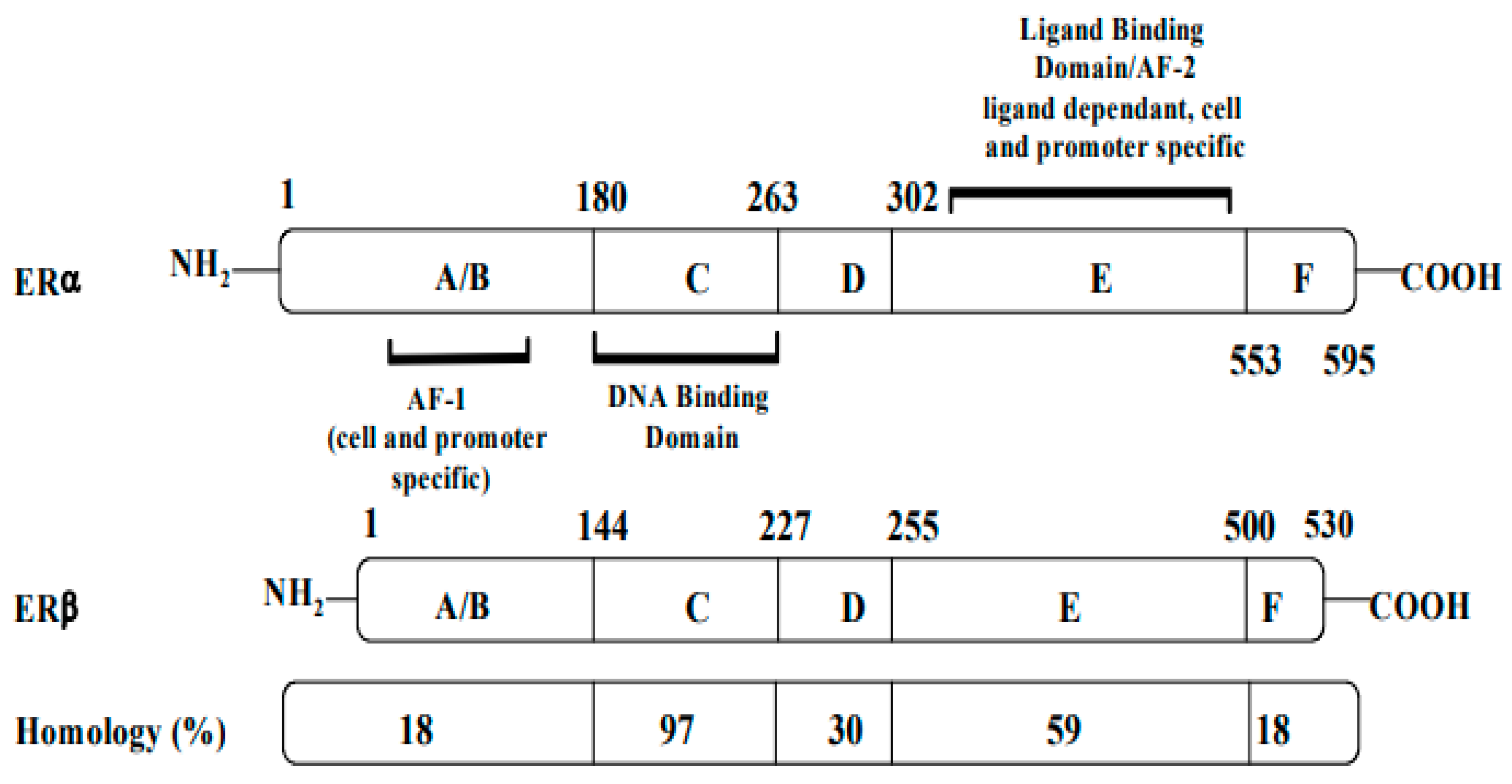
Figure 1. Structures, functional regions, and percentage of sequence homology of two human estrogen receptors: ERα and ERβ.
Both receptors are organized in six functional domains: the A/B domain at the N-terminal, the C domain of the DNA-binding domain (DBD), the D domain of the nuclear translocation signal, and the E/F domain at the C-terminus including the ligand-binding domain (LBD) and the ligand-dependent activation function AF-2.
AF: activation function (taken and modified from reference [2]).
Although the two ERs possess different functions, they share a homologous DNA-binding domain and a 55–60% homology in their ligand-binding domains (LBDs). Both coexist in breast tissues, with more ERα than ERβ in breast tumors [3]. While AF-1 has only 20% homology in ERα and ERβ, AF-2 is especially similar in both ERs [4].
In the presence of natural or synthetic agonists of the ER, AF-1 and AF-2 domains will interact with steroid receptor coactivator proteins. These include the p160/steroid receptor coactivator (SRC) family, which comprises three pleiotropic coregulators (SRC-1, SRC-2, and SRC-3) that serve as the transcription factors that regulate the expression of genes [5][6]. Hormone-activated receptors form multiple dimers of αα monomers, ββ monomers, and a combination of αβ monomers [7]. Depending on the composition of the dimers, cell homeostasis regulation and the affinity for different estrogen response elements (EREs) might vary.
The selective action of the ER might depend on the structure of the ligand, the type or isotype of ER, the type of co-regulator, and the interaction of the complex with EREs. Figure 2 depicts the action of ER in terms of explaining the tissue-specific binding of E2 to its receptors.
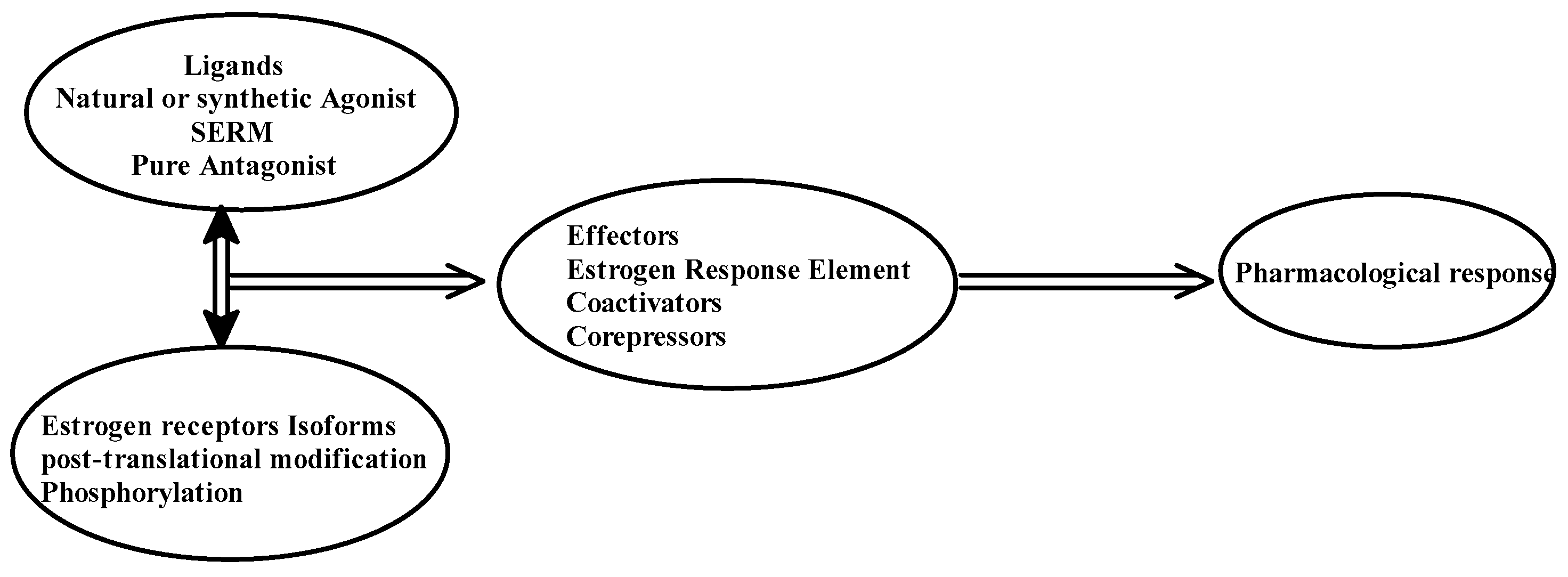
Figure 2. Pharmacological action of estrogen receptors.
ER ligands are classified into four families: ER agonists, SERDs, SERMs, and aromatase inhibitors (Figure 3). ER agonists include endogenous natural 17β-estradiol (Figure 3I), diethylstilbestrol (Figure 3II), and ethinylestradiol (Figure 3III). In addition, 17β-estradiol (E2) agonizes ERs in all tissue types where they are expressed and recruit coactivators to activate gene expression under the control of EREs, thereby promoting DNA synthesis and the proliferation of ER-responsive cells. Selective degraders of estrogen receptors (SERDs) completely inhibit and degrade ERα. Examples include the synthetic drugs ICI 182,780 (Fulvestrant) (Figure 3IV) and ICI 164,384 (Figure 3V), with the systematic name of N-n-butyl-N-methyl-11-[3,17β-dihydroxy-estra-1,3,5(10)-trien,7α-yl]-undecanamide. Selective modulators of estrogen receptors (SERMs) are unique in their anti-estrogen activity, depending on the type of tissue. Mounting evidence supports the unique characteristics of SERM activity, which is primarily determined by the selective recruitment of both ERα repressors and activators into specific tissues [4][8]. The main example is tamoxifen (TAM), which is a partial agonist of the first generation of SERMs (Figure 3VI). The final class of drugs, including anastrozole (Figure 3IX) and letrozole (Figure 3X), are aromatase inhibitors (AIs). AIs inhibit the aromatization of androgens at the CYP9A1 level, and block the endogenous formation of estrogens.
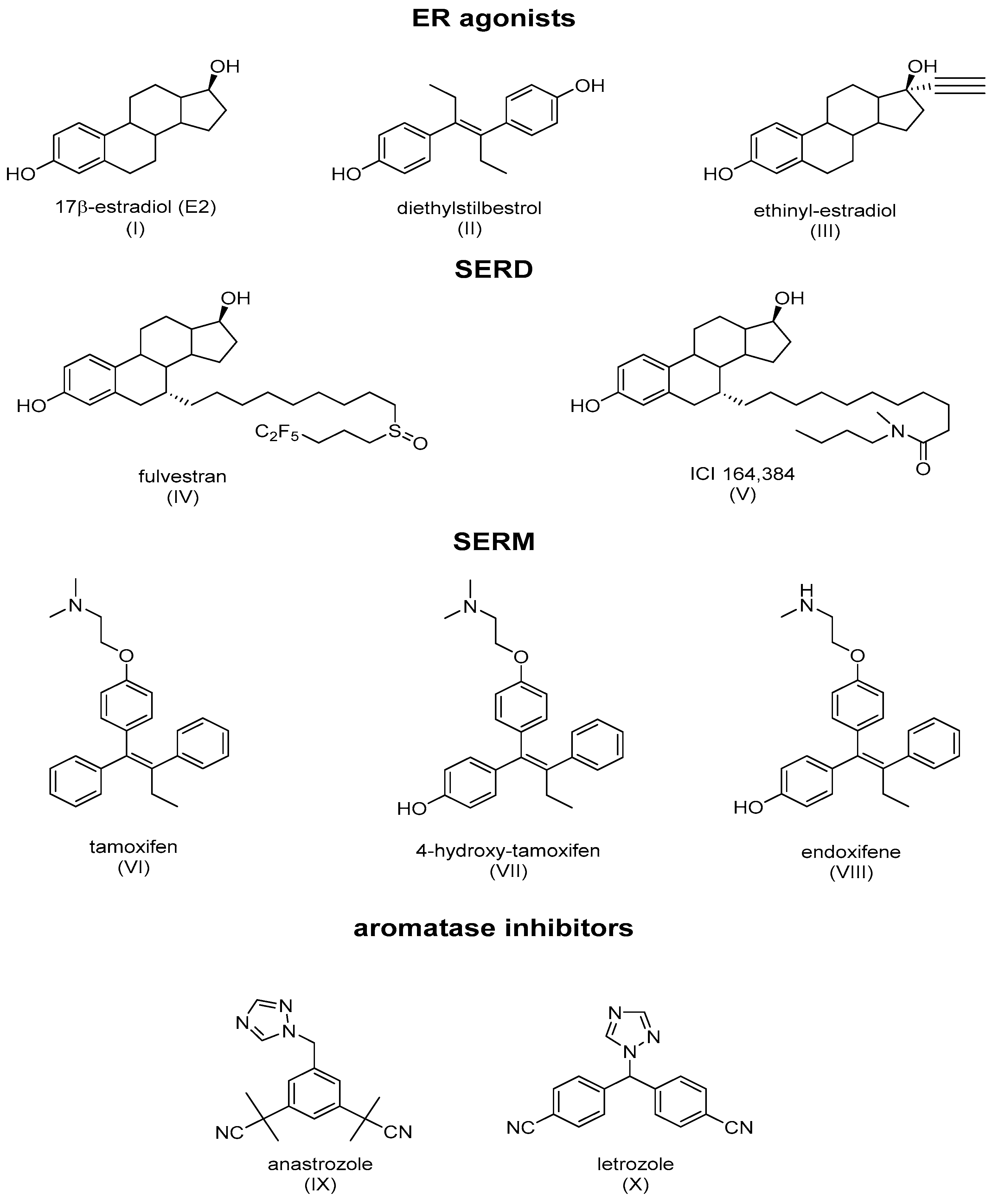
Figure 3. Main classes of agents used in hormone therapy that target estrogen signaling: ER agonists, SERDs (Fulvestran and ICI 164,384), SERMs (tamoxifen), and aromatase inhibitors.
TAM acts by blocking the mitogenic action of E2, via competition with E2 on ERα [9][10]. The structural analyses of ERα-4OHTam were conducted and showed that the drug effectively accommodates the ER ligand-binding pocket [11][12][13]. OH-TAM induced conformational changes that were different from E2 on ER-LBD, which affected the recruitment of the co-regulators and the regulation of gene expression [14]. The LBD, which consists of amino acids 304 to 553 of the ER, is composed of a cluster of twelve α-helices (H1–H12), which harbor a highly structured ligand-dependent domain containing dimerization interfaces, co-activation and co-repression functions, and a hydrophobic pocket that accommodates hormones.
ERα, in the absence of hormones, is inactivated as a complex by several chaperone proteins, such as HSP90, HSP70, and HSP40, with the aid of several other co-chaperones including cyclophilin-40 and p23 proteins. These heat shock proteins help to maintain the receptor in an adequate conformation, allowing for it to respond quickly to a hormonal signal. The binding of the hormone to the receptor results in the release of chaperonins and induces the nuclearization of the ER-E2 complex, which is followed by dimerization and stabilization in a conformation where the last helix (H12) folds over the ligand-binding pocket (LBP) and forms a hydrophobic groove, sealing the LBP [15].
In the presence of agonists, helix 12 traps the ligand inside the hydrophobic pocket of the ligand-binding domain. Helix 12’s hydrophobic grooves become exposed to the nuclear box recognition sequence containing the key LXXLL motif, which is rich in leucine. Alternatively, SERM drives the hydrophobic pocket into an open state, which dislocates helix 12 from forming an active AF-2 conformation, and specifically occupies the space for a coregulatory protein recognition sequence of LXXLL binding, thereby blocking the coactivators from binding and preventing gene transcription. The crystal structures of LBD complexed with 4-hydroxy-tamoxifen and ICI-164, 384 (Figure 3V) (pure antiestrogen) reveal evidence of certain critical parameters with regard to these interactions. Figure 4 depicts the mechanism of action of the ER ligands at the ERα level. E2, TAM, and ICI 164,384 (ICI) bind to ER LBD, which explains why they compete with E2 at the ERα level and can inhibit E2-mediated BC cell proliferation. TAM might exert its antiproliferative potency through other ER-dependent mitogenic pathways [16][17][18][19][20]. TAM also possesses ERα-independent antiproliferative properties and displays a certain degree of efficacy in the context of ER-negative cancer cells (see vide infra).
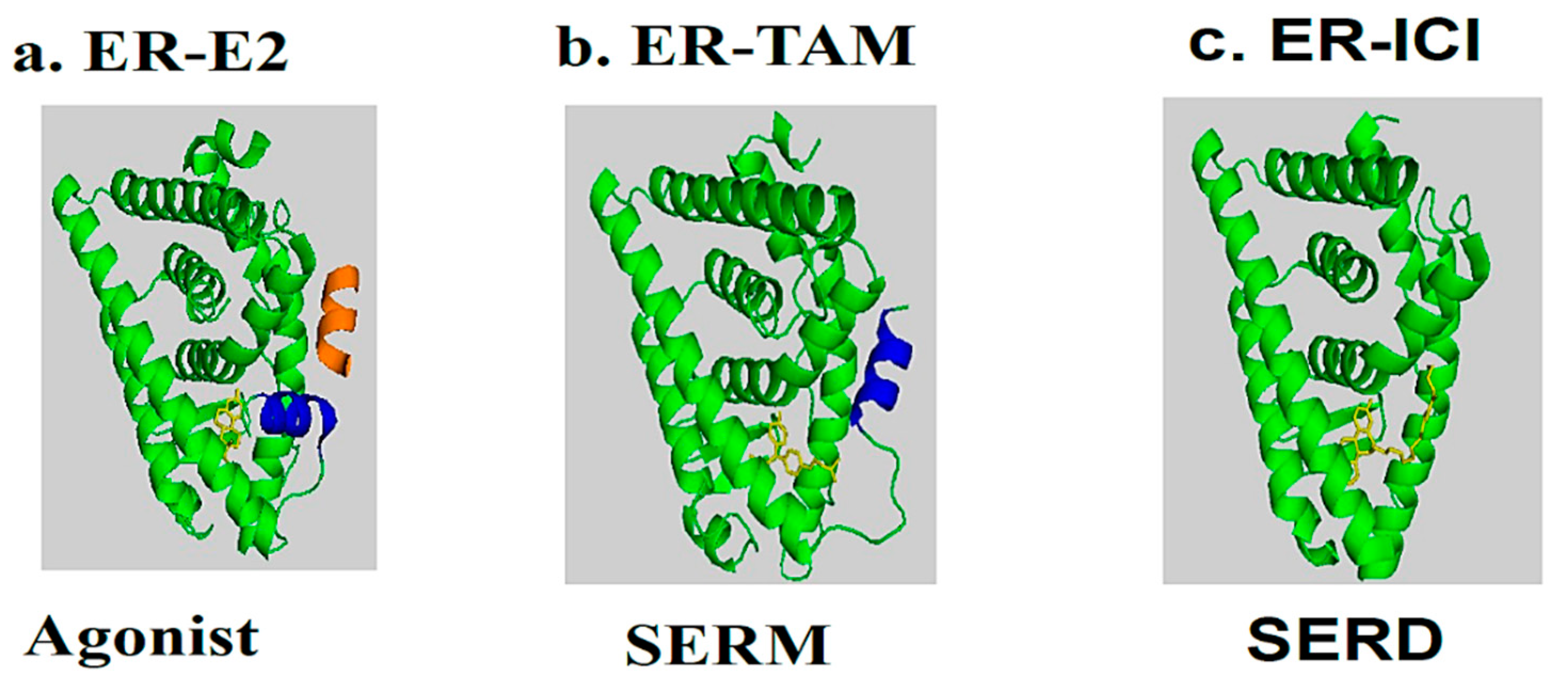
Figure 4. Molecular model and ribbon representation highlighting the action mechanism of estrogen receptors and the positioning of helix H12. (a) ER-E2 represents estradiol (yellow) in the presence of a co-activator (orange). (b) ER-TAM shows ER-LBD with tamoxifen (yellow), which is conducted to induce co-repression (SERM). (c) The ER-ICI interaction shows the full antagonist activity (SERD) in the absence of helix H12. Helix H12 in (a,b) is drawn as a cylinder in blue (Poirot et al., unpublished data).
2. Insight to the Chemo-Preventive Action of Tamoxifen in Breast Cancer
Chemo-prevention is an oncological prophylaxis that modulates the process of carcinogenesis in the early stages of cancer development with the use of pharmacological agents and non-nutritional food components [21]. Since the 1970s, TAM has been a landmark form of BC treatment. It was initially recommended as an antiestrogen to help treat hormone-responsive BC, but its curative potential was later expanded to adjuvant therapy in order to help reduce the incidence of BC. TAM inhibits cell proliferation through its actions on the ERα, which regulates the processing of growth factors. This can lead to the arrest of cell growth, or cell death and tumor regression. TAM was, therefore, shown to be an efficient chemo-preventive agent in both postmenopausal and premenopausal women with a moderate risk of BC [22]. The results of meta-analyses of randomized clinical trials and case–control studies reported a decrease in BC incidence in women receiving TAM as a postoperative adjuvant compared to untreated women [23][24][25][26][27][28][29][30][31].
Adjuvant TAM is commonly administered for 5 years; however, 10 years of TAM therapy has been associated with a greater efficacy in patients with ER-positive BC. In addition, this longer duration of therapy might reduce mortality caused by BC during the second decade after diagnosis by half [30][32]. Moreover, TAM reduces the risk of developing ER-positive BC by 48% in women over 35 years old [33]. Although the main rationale in the clinical use of TAM is the blockage of mitogenic actions of E2, several other mechanisms of TAM have been observed that could account, in part, to its anticancer action. Furthermore, TAM has been shown to induce cell death, which has been earlier summarized in certain other excellent reviews [34][35][36].
TAM binds with a nanomolar affinity to a microsomal binding site, called the anti-estrogen binding site (AEBS) [37]. Selective AEBS ligands include the diphenyl methane compounds PBPE (Figure 5I) and DPPE (tesmilifene, Figure 5II). AEBS ligands include SERMs, bearing a cationic amino alkyl side chain; estrogens and pure antiestrogens, on the other hand, have no affinity for the AEBS. Oxysterols (e.g., 6-ketocholestanol (Figure 5III), 7-ketocholestanol (Figure 5VI), and 7-ketocholesterol (Figure 5V), as well as unsaturated fatty acids and histamine), have also been characterized as AEBS ligands [35]. AEBS is found in various tissues, including BC cells, and is independent of ER status. Kedjouar et al. reported that AEBS is a multiproteic complex composed of two cholesterogenic enzymes: 3β-hydroxysterol-Δ8-Δ7-isomerase (EBP/D8D7I) and 3β-hydroxysterol-Δ7-reductase (DHCR7) [38]. It was later shown that the AEBS carries out cholesterol-5,6-epoxide hydrolase (ChEH) enzymatic activity that catalyzes the hydrolysis of 5,6-epoxycholestan-3β-ol diastereomers (5,6α-EC (Figure 5VI) and 5,6β-EC (Figure 5VII) into cholestane-3β,5α,6β-triol (CT) (Figure 5VIII) [39][40].
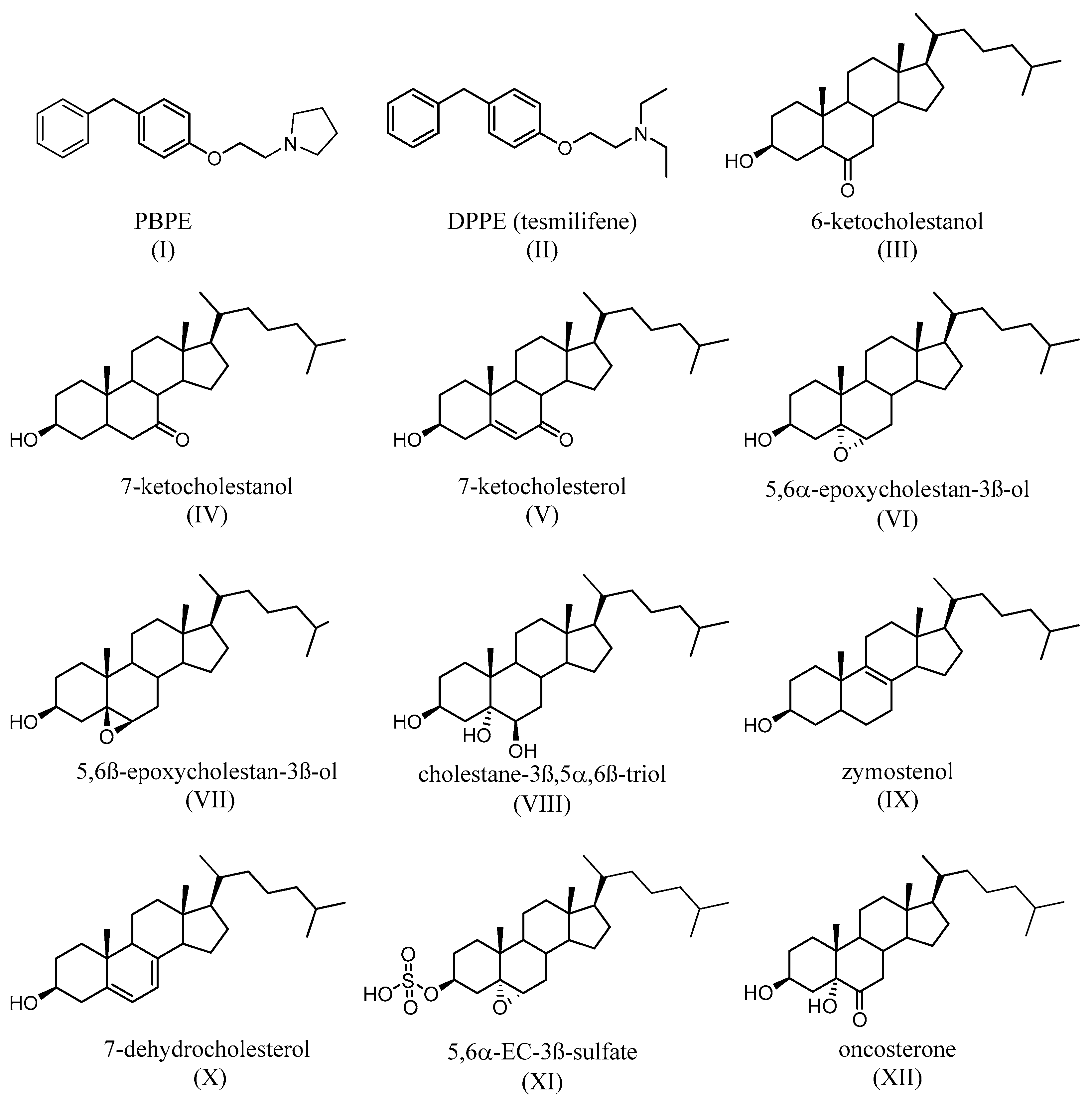
Figure 5. Synthetic (I,II) and endogenous (III–V) selective AEBS ligands with no affinity for ER. Oxysterols that accumulate after the inhibition of ChEH by AEBS ligands (VI–VII). Product of the ChEH enzymatic activity (VIII). Cholesterol precursors that can accumulate after the binding of ligands (including SERMs) on the AEBS (IX,X). The signaling sterol that accumulates in cells expresses SULT2B1b, after ChEH inhibition by AEBS ligands (XI). The tumor promoter whose biosynthesis is blocked by AEBS ligands (including SERMs) (XII).
TAM, SERMs, and the selective AEBS ligand PBPE (Figure 5I) induce the intracellular accumulation of cholesterol precursors, including zymostenol (Figure 5IX), the substrate of D8D7I, and 7-dehydrocholesterol (Figure 5X), which is the substrate of DHCR7. AEBS ligands also increase the total sterol levels in the cell [41]. Interestingly, accumulated cholesterol precursors undergo oxidation toward unidentified oxysterols. Since AEBS also carries out ChEH activity, the intracellular accumulation of 5,6-EC and the diminution of CT (Figure 5VIII) have thus been described, on BC cells, as being exposed to AEBS ligands. In addition, CT is metabolized by 11β-hydroxysteroid-dehydrogenase type 2 into oncosterone (Figure 5XII), an oncometabolite that promotes BC development [42]. Consequently, targeting ChEH with AEBS ligands inhibits both the production of CT and oncosterone in BC cells. It was shown that the inhibition of oncosterone production represents a promising approach for the development of new anticancer agents [42][43][44].
3. Acquired Resistance to Tamoxifen
Unfortunately, within a few years, almost all responsive patients eventually develop acquired resistance [45]. The development of resistance to the effects of drugs remains a major obstacle in hormonal therapy with TAM. Resistance to the effects of TAM might be the result of various mechanisms that might be linked to high levels, losses, or alterations in the ER. A total of 30% of ER-positive tumors acquire resistance to TAM [46]. Compared to their more treatment-resistant HER2-positive counterparts, ER-negative and -positive patients responded well to treatment with TAM, regardless of their progesterone status [47]. Additionally, the ER can be active in cell membranes. When bound to estrogen or SERMs, it could trigger cellular proliferation, where TAM acts as an agonist [48][49]. An example of a mechanism underlying resistance is the intricate interaction of surface receptors, especially G-protein-coupled receptors (GPCRs) [50]. It has been demonstrated that G-protein-coupled estrogen receptors (GPERs) mediate multiple estrogenic signals in various types of malignant cells [51] and respond to estrogen via the up-regulation of aromatase (also called estrogen synthetase or estrogen synthase, CYP19A1), the adrenal enzyme that converts androstenedione and estrone to estrogen (T) [52]. More particularly, GPR30 was detected in nearly 62% of invasive tumors and was co-expressed with the ER in about 43% of BC cases. Furthermore, the expression of GPR30 is inversely correlated with the expression of ER [53] and also attenuates the inhibition of mitogen-activated protein kinases (MAP kinases), thereby contributing to resistance to TAM in BC [54][55]. Resistance to TAM might also take place through growth factors, survival factors, and chemokines, which support tumorigenesis. Such signaling, which includes HER-2/neu, TGFβ, Notch, and the insulin-like growth factor receptor, demonstrates multiple pathophysiological functions associated with oncogenic kinase signal transduction pathways, such as the phosphatidylinositol3-kinase (PI3K)/Akt/mammalian target of the rapamycin (mTOR) signaling pathway (PI3K/Akt/mTOR) and the Ras/Raf/MEK/ERK axis [48][56][57][58][59][60][61][62][63][64].
In breast cancer cells, TAM up-regulates mRNA for transforming growth factor-beta (TGF-β2) with no correlation with the status of αER and PR [57]. TGF-β2 is a secreted protein known as a cytokine that can be involved in various cellular functions. Specifically, in BC, TAM inhibits the TGF-β-mediated activation of breast fibroblasts. TAM blocks non-SMAD signaling through ERK1/2 MAP-kinase and the transcription factor FRA2 [64]. Therefore, TAM might provide therapeutic benefits by inhibiting the differentiation of myofibroblasts. Myofibroblasts are involved in promoting the growth and invasiveness of tumors [65]. However, the differentiation and activation of myofibroblasts have been observed in other cancers, such as pancreatic cancer, where they contribute to the prevention of the development of tumors; thus, the antimyofibroblast effect of TAM might cause chemoresistance in BC [66]. Moreover, TGFβ might also promote epithelial-mesenchymal transition (EMT) and cell invasiveness, depending on the cell context, growth factor environment, and stage of BC [67]. The delineation of the other molecular aspects associated with resistance to the therapeutic effects of TAM includes impaired metabolism, especially by CYP2D6 [68]. Certain CYP2D6 variants were reported to diminish clinical outcomes in patients treated with TAM [69][70].
The effect of TAM on metabolism involving glucose is another important mediator in the resistance of cancer cells to TAM. Glucose transporter 1 (GLUT1) plays a role in increasing autophagy and in acquiring resistance to TAM in BC cells [71]. In fact, the overexpression of GLUT1 has been observed in aggressive breast cancer and is correlated with a poor prognosis for BC patients [72]. During the development of resistance of BC cells to TAM, the activation of protein kinase B, also known as AKT kinase, and the decreased levels of AMPK lead to the activation of hypoxia-inducible factor 1α (HIF-1α). AKT is the collective name for a set of three serine/threonine-specific protein kinases that are involved in various processes in cells, including glucose metabolism, apoptosis, proliferation, transcription, and the migration of cells. The development of resistance to TAM was found to be driven by hypoxia-inducible factor-1 alpha HIF-1α, making the AKT and AMPK pathways good targets for overcoming resistance to TAM [73]. (HIF-1α) regulates the responses of cells to oxygen concentration, supporting the adaptation of tumor cells to hypoxia in the oxygen-deficient microenvironment of rapidly growing tumors. Thus, HIF-1α is important in carcinogenesis and progression tumors. HIF-1α is associated with a poor prognosis in BC patients [74].
TAM accumulates in the mitochondria and can affect multiple mitochondrial functions. TAM inhibits oxidative phosphorylation and fatty acid oxidation by binding to the Flavin mononucleotide molecule of complex-I, thus resulting in mitochondrial electron transport chain dysfunction [75]. Cells resistant to TAM are characterized by greater glycolysis and exhibit increased AMPK phosphorylation and the activation of mitochondrial protein deacetylase, which is known as Sirtuin 3 (SIRT3) [76][77].
TAM might behave as an estrogen agonist in BC cells that express greater amounts of human epidermal growth factor receptor 2 (HER2), a protein that promotes the growth of cancer cells, resulting in de novo resistance [78]. TAM might bind and activate ERα36—a variant of ERα in BC stem cells that is associated with poor prognoses—in order to enhance the stemness and metastasis of BC cells via the transcriptional stimulation of the protein-coding gene Aldehyde Dehydrogenase 1 Family Member A1 (ALDH1A1) [79]. In a more recent report, it was reported that oncogenic p21-activated kinase-1 (PAK1) might also reload resistance to TAM by phosphorylating ERα and other substrates in ER-positive BC patients [80]. Alternative mechanisms of resistance to TAM might also be due to the activation of inflammatory cytokines, such as interleukin-1 beta (IL-1β). IL1β is also known as leukocytic pyrogen, leukocytic endogenous mediator, mononuclear cell factor, and lymphocyte activating factor. Resistance to TAM might also be caused by mechanisms involving tumor necrosis factor and alpha (TNFα), which are associated with Nuclear Factor-Kappa B (NF-κB) [81].
BC inhibition might be mediated by nuclear factor erythroid-2 related factor-2 (Nrf2), which is a member of the cap ‘n’ collar (CNC) subfamily of the basic region leucine zipper (bZip) transcription factor. This acts as the master regulator controlling the expression of antioxidant genes that regulate physiological and pathophysiological events following exposure to oxidants. Under oxidative stress, the complex Kelch-like ECH-associated protein 1 (KEAP1), which is a subunit of CULLIN 3 (CUL3)-based E3 ubiquitin ligase, and Nrf2 dissociates, and Nrf2 is then translocated into the nucleus where it heterodimerizes with the small musculoaponeurotic fibrosarcoma proteins (MAF), which are basic region leucine zipper-type transcription factors that can bind to DNA and regulate gene regulation by binding to the antioxidant-responsive element (ARE) and stimulating the gene expression of antioxidants and detoxification enzymes. Consequently, the knockdown of Nrf2 increases TAM-induced cell death in TAM-resistant cells [82][83][84][85][86]. MicroRNAs (miRNAs), a class of short non-coding RNAs commonly involved in treatment regimens, were shown to play a pivotal role as regulators of various biological processes, such as the therapeutic targets involved in ERα regulation [85]. The mechanisms of resistance have been correlated with a change in the expression of miRNA, as well as the remodeling of the epithelial to mesenchymal transition (EMT) which is involved in the formation of tumors [86]. The expression of miR-221 and miR-222, encoded in tandem on the X chromosome, has been found to be elevated two-fold in endocrine-therapy-resistant HER2/neu-positive primary human BC tissues [87][88]. Although TAM is involved in the regulation of microRNAs, miR-221/222 has been found to confer resistance to TAM in BC by reducing the expression of the cell cycle regulator p27/kip1 [89].
Another possible contributor to the resistance is the microsomal antiestrogen binding site (AEBS). The ligands of the AEBS do not bind to ERs, but bind selective ER modulators (SERMs). AEBS ligands contain a hydrophobic core that mimics the steroid backbone of estrogens. The antitumor properties of AEBS ligands and their relationship with cholesterol metabolism perturbations have been discussed previously. AEBS ligands, including tesmilifene (Figure 5II) and PBPE (Figure 5I), display antitumor properties through their impact on cholesterol metabolism. Indeed, AEBS ligands, including SERMs, induce the redifferentiation of BC cells into lactating epithelial cells, which is characterized by a cell cycle arrest in the G0-G1 phase, morphological modifications of triglycerides, and the secretion of milk proteins [42][90][91].
TAM and N-pyrrolidino- (phenylmethyphenoxy)-ethanamine,HCl) (PBPE) trigger intracellular formation and the accumulation of 5,6α-EC and 5,6β-EC in BC cells through both the induction of oxidative stress and the inhibition of ChEH. In addition, 5,6α-EC is transformed into CES (Figure 5XI) in MCF-7 cells that express SULT2B1b but not in MDA-MB-231 cells that do not express SULT2B1b. Both 5,6α-EC and CES are involved in triglyceride accumulation (a characteristic of BC cell differentiation) and in cell death through an LXRβ-dependent mechanism. It is noteworthy that both 5,6α-EC and CES have been previously reported to be LXRβ modulators [44][92]. Furthermore, 5,6β-EC also participates in cell death induction through an LXRβ-independent mechanism. In contrast to sensitive MCF7 cells, MDA-MB-231 is resistant to the TAM inhibition of E2 mitogenic activity. Importantly, the ectopic expression of SULT2B1b in MDA-MB-231 cells increases the sensitivities of these cells with respect to the cytotoxic activity of TAM and the selective AEBS ligand, PBPE. In addition, MDA-MB-231 cells are as sensitive as MCF7 to the cytotoxic activity of CES (Figure 5XXI).
Altogether, TAM’s impact on cholesterol metabolism via the AEBS/ChEH/Sult2B1b/LXRβ pathway is a key mediator of the sensitivity and resistance of BC. The roles of several metabolites (5,6-EC, CES, and oncosterone), related enzymes (D8D7I, DHCR7, SULT2B1b, and 11β-HSD2), oxidative stress/antioxidant defenses (NRF2, SOD, Catalase, NADPH, and GSH), and nuclear receptors (LXR) deserve to be considered for the purposes of a better understanding of the intrinsic and acquired mechanism of resistance against TAM. The relationship between the ER status of BC and these metabolic pathways also remains unanswered. These events might convert ER-dependent BCs to hormone-independent human BC cells and thus lead to resistance to TAM.
4. Side Effects of Tamoxifen
The hormonal treatment of breast cancer is a procedure that drastically affects a patient’s quality of life. Due to complex drug regimens that are conducted in order to treat comorbidity conditions, older patients are more prone to adjuvant hormone intolerance [93]. Statistically, the tolerance toward hormone therapy among older BC patients ranges between 41% and 72% [94]. During greater doses of TAM, reactive oxidative species (ROS) are produced, which can have deleterious effects in not only BC cells, but also healthy cells. As a result, the release of ROS might lead to unintentional adverse effects under long-term therapy, including high toxicity and genotoxicity [24]. TAM induces a myriad of side effects, which include endometrial hyperplasia, polyps, fibrosis, cystic atrophy, and uterine sarcoma [95][96]. In a clinical study conducted on 204 patients aged between 27 and 84, after a 5-year period of TAM treatment, thromboembolic and uterine cancer mortality were only observed in women older than 55 [23]. In another random-effect meta-analysis report involving 53,000 women, the risk of endometrial cancer and of vascular and thrombotic events appeared to be less in premenopausal women [97]. Consequently, TAM did not increase the risk of endometrial cancer in premenopausal patients [98].
Patients might also experience cognitive disturbances known as TAM brain fog, which can result in effects such as decision-making impairment and the deterioration of executive functions [99][100], as well as depression [101]. Other complications encompass arthralgia, chills, night sweats, irregular heartbeat, insomnia, and hot flashes [102][103][104][105][106][107]. The long-term use of TAM might cause steatogenic hepatotoxicity in mice [108] and rats. In addition, TAM induced a liver iron overload, with unaltered hepatic function, in non-diabetic rats and might be a useful tool for investigating the biological control of iron metabolism [109]. The International Agency for Research on Cancer (IARC) rates TAM as being carcinogenic in experimental animals [110]. In a cross-sectional study of 32 women who were given TAM compared to 39 control women, TAM was reported to increase steatosis and adipose tissue distribution through its anti-estrogenic effect [111]. Massive hepatic steatosis in premenopausal women was also observed [112], suggesting that functional polymorphism in CYP17 is associated with circulating estrogen levels and is involved in basal hepatic lipid metabolism. An unsuitable effect of TAM on lipid metabolism includes hypertriglyceridemia [113][114], as well as severe acute pancreatitis in a dose-dependent manner [115]. Treatment with TAM was also correlated with clinical manifestations in the form of abnormal spotting or bleeding [96]. Other complications include congestion and sexual dysfunction [116]. In addition, gastrointestinal cancers [117] were also mentioned.
References
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor alpha and beta: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402.
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during ageing: From periphery to brain Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209.
- Saji, S.; Hirose, M.; Toi, M. Clinical significance of estrogen receptor in breast cancer. Cancer Chemother. Pharmacol. 2005, 1, 21–26.
- Maximov, P.Y.; Lee, T.M.; Jordan, V.C. Discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr. Clin. Pharmacol. 2013, 8, 135–155.
- Torchia, J.; Rose, D.W.; Inostroza, J.; Kamei, Y.; Westin, S.; Glass, C.K.; Rosenfeld, M.G. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 1997, 387, 677–684.
- Musa, M.; Khan, M.O.; Copperwood, J.S. Medicinal chemistry and emerging strategies applied to the development of selective estrogen receptor modulators (SERMs). Curr. Med. Chem. 2007, 14, 1249–1261.
- Shaaban, A.M.; Green, A.R.; Karthik, S.; Alizadeh, Y.; Hughes, T.A.; Harkins, L.; Ellis, I.O.; Robertson, J.F.; Paish, E.C.; Saunders, P.T.K.; et al. Nuclear and cytoplasmic expression of ERbeta1, ERbeta2, and ERbeta5 identifies distinct prognostic outcome for breast cancer patients. Clin. Cancer Res. 2008, 14, 5228–5235.
- Lewis, J.S.; Jordan, V.C. Selective estrogen receptor modulators (SERMs): Anticancer and drug resistance mechanisms. Muta. Res. 2005, 591, 247–263.
- Jordan, V.C. Tamoxifen: A most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003, 2, 205–213.
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658.
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758.
- Pike, A.C.; Brzozowski, A.M.; Hubbard, R.E.; Bonn, T.; Thorsell, A.G.; Engstrom, O.; Ljunggren, J.; Gustafsson, J.A.; Carlquist, M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999, 18, 4608–4618.
- Pike, A.C.; Brzozowski, A.M.; Walton, J.; Hubbard, R.E.; Thorsell, A.G.; Li, Y.L.; Gustafsson, J.A.; Carlquist, M. Structural insights into the mode of action of a pure antiestrogen. Structure 2001, 9, 145–153.
- Thomas, C.; Gustafsson, J.A. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608.
- Rosano, C.; Stec-Martyna, E.; Lappano, R.; Maggiolini, M. A structure-based approach for the discovery of new selective estrogen receptor modulators. Curr. Med. Chem. 2011, 18, 1188–1194.
- Webb, P.; Lopez, G.N.; Uht, R.M.; Kusher, P.J. Tamoxifen activation of the estrogen receptor/AP-1 pathway: Potential origin for the cell specific estrogen-like effects of antiestrogen. Mol. Endocrinol. 1995, 9, 443–456.
- Clarke, R.B.; Anderson, E.; Howell, A. Steroid receptors in human breast cancer. Trends Endocrinol. Metab. 2004, 15, 316–323.
- Fox, E.M.; Andrade, J.; Shupnik, M.A. Novel actions of estrogen to promote proliferation: Integration of cytoplasmic and nuclear pathways. Steroids 2009, 74, 622–627.
- Doisneau-Sixou, S.F.; Sergio, C.M.; Carroll, J.S.; Hui, R.; Musgrove, E.A.; Sutherland, R.L. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr. Relat. Cancer 2003, 10, 179–186.
- Peyrat, J.P.; Bonneterre, J. Type 1 IGF receptor in human breast diseases. Breast Cancer Res. Treat. 1992, 22, 59–67.
- Baer-Dubowska, W.; Ignatowicz, E. Chemoprevention of Cancer Basic Mechanisms and Molecular Targets. In Carcinogenic and Anticarcinogenic food Components; Baer Dubowska, W., Bartoszek, A., Malejka-Giganti, D., Eds.; CRC Taylor&Francis Group: Boca Raton, FL, USA; London, UK, 2006; pp. 177–196. ISBN 978-0849320965.
- Nelson, H.D.; Fu, R.; Griffin, J.C.; Nygren, P.; Smith, M.E.; Humphrey, L. Systematic review: Comparative effectiveness of medications to reduce risk for primary breast cancer. Ann. Intern. Med. 2009, 151, 703–715.
- Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Pan, H.C.; Taylor, C.; Wang, Y.C.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomized trials. Lancet 2011, 27, 771–778.
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Redmond, C.K.; Kavanah, M.; Cronin, W.M.; Vogel, V.; Robidoux, A.; Dimitrov, N.; Atkins, J.; et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer Inst. 1998, 90, 1371–1388.
- Coombes, R.C.; Hall, E.; Gibson, L.J.; Paridaens, R.; Jassem, J.; Delozier, T.; Jones, S.E.; Alvarez, I.; Bertelli, G.; Ortmann, O.; et al. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N. Engl. J. Med. 2004, 350, 1081–1092.
- Cuzick, J.; Forbes, J.; Edwards, R. First results from the International Breast Cancer Intervention Study (IBIS-I): A randomised prevention trial. Lancet 2002, 360, 817–824.
- Cuzick, J.; Powles, T.; Veronesi, U.; Forbes, J.; Edwards, R.; Ashley, S.; Boyle, P. Overview of the main outcomes in breast-cancer prevention trials. Lancet 2003, 361, 296–300.
- Cuzick, J.; Forbes, J.F.; Sestak, I.; Cawthorn, S.; Hamed, H.; Holli, K.; Howell, A. Long-term results of tamoxifen prophylaxis for breast cancer—96-month follow-up of the randomized IBIS-I trial. J. Natl. Cancer Inst. 2007, 99, 272–282.
- Swaby, R.F.; Sharma, C.G.; Jordan, V.C. SERMs for the treatment and prevention of breast cancer. Rev. Endocr. Metab. Disord. 2007, 8, 229–239.
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Alencar, V.H.M.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptorpositive breast cancer ATLAS a randomised trial. Lancet 2013, 381, 805–816.
- Wilkes, G.M.; Barton-Burke, M. 2020–2021 Oncology Nursing Drug Handbook; Jones & Bartlett Learning: Burlington, NJ, USA, 2019; ISBN 13: 978-1284171327.
- Parton, M.; Smith, I.E. Controversies in the management of patients with Breast cancer: Adjuvant endocrine therapy in premenopausal women. J. Clin. Oncol. 2008, 26, 745–752.
- Kramer, R.; Brown, P. Should tamoxifen be used in breast cancer prevention? Drug Saf. 2004, 27, 979–989.
- de Médina, P.; Favre, G.; Poirot, M. Multiple targeting by the antitumor drug tamoxifen: A structure-activity study. Curr. Med. Chem. Anti-Cancer Agents. 2004, 4, 491–508.
- Leignadier, J.; Dalenc, F.; Poirot, M.; Silvente-Poirot, S. Improving the efficacy of hormone therapy in breast cancer: The role of cholesterol metabolism in SERM-mediated autophagy, cell differentiation and death. Biochem. Pharmacol. 2017, 144, 18–28.
- Bogush, T.A.; Polezhaev, B.B.; Mamichev, I.A.; Bogush, E.A.; Polotsky, B.E.; Tjulandin, S.A.; Ryabov, A.B. Tamoxifen Never Ceases to Amaze: New Findings on Non-Estrogen Receptor Molecular Targets and Mediated Effects. Cancer Investig. 2018, 36, 211–220.
- Kedjouar, B.; Daunes, S.; Vilner, B.J.; Bowen, W.D.; Klaebe, A.; Faye, J.C.; Poirot, M. Structural similitudes between cytotoxic antiestrogen-binding site (AEBS) ligands and cytotoxic sigma receptor ligands. Evidence for a relationship between cytotoxicity and affinity for AEBS or sigma-2 receptor but not for sigma-1 receptor. Biochem. Pharmacol. 1999, 58, 1927–1939.
- Kedjouar, B.; de Médina, P.; Oulad-Abdelghani, M.; Payré, B.; Silvente-Poirot, S.; Favre, G.; Faye, J.C.; Poirot, M. Molecular characterization of the microsomal tamoxifen binding site. J. Biol. Chem. 2004, 279, 34048–34061.
- de Medina, P.; Paillasse, M.R.; Segala, G.; Poirot, M.; Silvente-Poirot, S. Identification and pharmacological characterization of cholesterol-5,6-epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 13520–13525.
- Silvente-Poirot, S.; Poirot, M. Cholesterol epoxide hydrolase and cancer. Curr. Opin. Pharmacol. 2012, 6, 696–703.
- Silvente-Poirot, S.; Poirot, M. Cancer Cholesterol and cancer in the balance. Science 2014, 343, 1445–1446.
- Voisin, M.; de Medina, P.; Mallinger, A.; Dalenc, F.; Huc-Claustre, E.; Leignadier, J.; Serhan, N.; Soules, R.; Segale, G.; Mougel, A.; et al. Identification of a tumor-promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2017, 114, E9346–E9355.
- de Medina, P.; Diallo, K.; Huc-Claustre, E.; Attia, M.; Soulès, R.; Silvente-Poirot, S.; Poirot, M. The 5,6-epoxycholesterol metabolic pathway in breast cancer: Emergence of new pharmacological targets. Br. J. Pharmacol. 2021, 178, 3248–3260.
- Poirot, M.; Mallinger, A.; Dalenc, F.; Soulès, R.; Silvente-Poirot, S. Chemistry, biochemistry, metabolic fate and mechanism of action of 6-oxo-cholestan-3β,5α-diol (OCDO), a tumor promoter and cholesterol metabolite. Biochimie 2018, 153, 139–149.
- Nielsen, K.V.; Ejlertsen, B.; Müller, S.; Møller, S.; Rasmussen, B.B.; Balslev, E.; Lænkholm, A.V.; Christiansen, P.; Mouridsen, H.T. Amplification of ESR1 might predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res. Treat. 2011, 127, 345–355.
- Rodriguez, D.; Ramkairsingh, M.; Lin, X.; Kapoor, A.; Major, P.; Tang, D. The central contributions of breast cancer stem cells in developing resistance to endocrine therapy in Estrogen Receptor (ER)-positive breast cancer. Cancers 2019, 11, 1028.
- Dowsett, M.; Houghton, J.; Iden, C.; Salter, J.; Farndon, J.; A’Hern, R.; Sainsbury, R.; Baum, M. Benefit from adjuvant tamoxifen therapy in primary breast cancer patients according estrogen receptor, progesterone receptor, EGF receptor and HER2 status. Ann. Oncol. 2006, 17, 818–826.
- Osborne, C.K.; Schiff, R. Growth factor receptor cross-talk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast 2003, 12, 362–367.
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr. Rev. 2008, 29, 217–233.
- Gaudet, H.M.; Cheng, S.B.; Christensen, E.M.; Filardo, E.J. The G-protein coupled estrogen receptor, GPER: The inside and inside-out story. Mol. Cell Endocrinol. 2015, 418, 207–219.
- Ignatov, T.; Claus, M.; Nass, N.; Haybaeck, J.; Seifert, B.; Kalinski, T.; Ortmann, O.; Ignatov, A. G-protein-coupled estrogen receptor GPER-1 expression in hormone receptor-positive breast cancer is associated with poor benefit of tamoxifen. Breast Cancer Res. Treat. 2019, 174, 121–127.
- Catalano, S.; Giordano, C.; Panza, S.; Chemi, F.; Bonofiglio, D.; Lanzino, M.; Rizza, P.; Romeo, F.; Fuqua, S.A.; Maggiolini, M.; et al. Tamoxifen through GPER upregulates aromatase expression: A novel mechanism sustaining tamoxifen-resistant breast cancer cell growth. Breast Cancer Res. Treat. 2014, 146, 273–285.
- Arias-Pulido, H.; Royce, M.; Gong, Y.; Joste, N.; Lomo, L.; Lee, S.J.; Chaher, N.; Verschraegen, C.; Lara, J.; Prossnitz, E.R.; et al. GPR30 and estrogen receptor expression: New insights into hormone dependence of inflammatory breast cancer. Breast Cancer Res. Treat. 2010, 123, 51–58.
- Filardo, E.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006, 12, 6359–6366.
- Yu, T.; Cheng, H.; Ding, Z.; Wang, Z.; Zhou, L.; Zhao, P.; Tan, S.; Xu, X.; Huang, X.; Liu, M.; et al. GPER mediates decreased chemosensitivity via regulation of ABCG2 expression and localization in tamoxifen-resistant breast cancer cells. Mol. Cell. Endocrinol. 2020, 506, 110762.
- Butta, A.; MacLennan, K.; Flanders, K.C.; Sacks, N.P.; Smith, I.; McKinna, A.; Dowsett, M.; Wakefield, L.M.; Sporn, M.B.; Baum, M. Induction of transforming growth factor beta 1 in human breast cancer in vivo following tamoxifen treatment. Cancer Res. 1992, 52, 4261–4264.
- Brandt, S.; Kopp, A.; Grage, B.; Knabbe, C. Effects of tamoxifen on transcriptional level of transforming growth factor beta (TGF-beta) isoforms 1 and 2 in tumor tissue during primary treatment of patients with breast cancer. Anticancer Res. 2003, 23, 223–229.
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Mohsin, S.K.; Osborne, C.K. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res. 2004, 10, 331S–336S.
- Rizzo, P.; Miao, H.; D’Souza, G.; Osipo, C.; Song, L.L.; Yun, J.; Zhao, H.; Mascarenhas, J.; Wyatt, D.; Antico, G.; et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008, 68, 5226–5235.
- McGlynn, L.M.; Kirkegaard, T.; Edwards, J.; Tovey, S.; Cameron, D.; Twelves, C.; Bartlett, J.M.; Cooke, T.G. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin. Cancer Res. 2009, 15, 1487–1495.
- Meyer, D.S.; Bentires-Alj, M. Can phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibition ERase them all. Breast Cancer Res. 2010, 12, 315.
- Tryfonidis, K.; Zardavas, D.; Katzenellenbogen, B.S.; Piccart, M. Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat. Rev. 2016, 50, 68–81.
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist 2018, 23, 528–539.
- Viedma-Rodríguez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F.; Diaz-Zaragoza, M.; Martínez-Hernández, G.; Ruiz Esparza-Garrido, R.; Velázquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer. Oncol. Rep. 2014, 32, 3–15.
- Carthy, J.M.; Sundqvist, A.; Heldin, A.; van Dam, H.; Kletsas, D.; Heldin, C.H.; Moustakas, A. Tamoxifen Inhibits TGF-β-Mediated Activation of Myofibroblasts by Blocking Non-Smad Signaling Through ERK1/2. J. Cell Physiol. 2015, 230, 3084–3092.
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238.
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014, 25, 719–734.
- Massague, J. TGFbeta in cancer. Cell 2008, 134, 215–230.
- Stearns, V.; Rae, J.M. Pharmacogenetics and breast cancer endocrine therapy: CYP2D6 as a predictive factor for tamoxifen metabolism and drug response. Expert. Rev. Mol. Med. 2008, 10, e34.
- Brauch, H.; Schwab, M. Prediction of tamoxifen outcome by genetic variation of CYP2D6 in post-menopausal women with early breast cancer. Br. J. Clin. Pharmacol. 2014, 77, 695–703.
- Goetz, M.P.; Sangkuhl, K.; Guchelaar, H.J.; Schwab, M.; Province, M.; Whirl-Carrillo, M.; Symmans, W.F.; McLeod, H.L.; Ratain, M.J.; Zembutsu, H.; et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for CYP2D6 and tamoxifen therapy. Clin. Pharmacol. Ther. 2018, 103, 770–777.
- Sun, M.; Zhao, S.; Duan, Y.; Ma, Y.; Wang, Y.; Ji, H.; Zhang, Q. GLUT1 participates in tamoxifen resistance in breast cancer cells through autophagy regulation Naunyn Schmiedebergs. Arch. Pharmacol. 2021, 394, 205–216.
- Kang, S.S.; Chun, Y.K.; Hur, M.H.; Lee, H.K.; Kim, Y.J.; Hong, S.R.; Lee, J.H.; Lee, S.G.; Park, Y.K. Clinical significance of glucose transporter 1 (GLUT1) expression in human breast carcinoma. Jpn. J. Cancer Res. 2002, 93, 1123–1128.
- Woo, Y.M.; Shin, Y.; Lee, E.J.; Lee, S.; Jeong, S.H.; Kong, H.K.; Park, E.Y.; Kim, H.K.; Han, J.; Chang, M.; et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1α Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0132285.
- Das, C.K.; Parekh, A.; Parida, P.K.; Bhutia, S.K.; Mandal, M. Lactate dehydrogenase A regulates autophagy and tamoxifen resistance in breast cancer. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1004–1018.
- Larosche, I.; Lettéron, P.; Fromenty, B.; Vadrot, N.; Abbey-Toby, A.; Feldmann, G.; Pessayre, D.; Mansouri, A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J. Pharmacol. Exp. Ther. 2007, 321, 526–535.
- Tomková, V.; Sandoval-Acuña, C.; Torrealba, N.; Truksa, J. Mitochondrial fragmentation, elevated mitochondrial superoxide and respiratory supercomplexes disassembly is connected with the tamoxifen-resistant phenotype of breast cancer cells. Free Radic. Biol. Med. 2019, 143, 510–521.
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 2007, 27, 8807–8814.
- Shou, J.; Massarweh, S.; Osborne, C.K.; Wakeling, A.E.; Ali, S.; Weiss, H.; Schiff, R. Mechanisms of tamoxifen resistance: Increased estrogen receptor-HER2/ neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004, 96, 926–935.
- Wang, Q.; Jiang, J.; Ying, G.; Xie, X.Q.; Zhang, X.; Xu, W.; Zhang, X.; Song, E.; Bu, H.; Ping, Y.F.; et al. Tamoxifen enhances stemness and promotes metastasis of ERα36+ breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res. 2018, 28, 336–358.
- Rajendran, S.; Swaroop, S.S.; Roy, J.; Inemai, E.; Murugan, S.; Rayala, S.K.; Venkatraman, G. p21 activated kinase-1 and tamoxifen—A deadly nexus impacting breast cancer outcomes. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188668.
- Stender, J.D.; Nwachukwu, J.C.; Kastrati, I.; Kim, Y.; Strid, T.; Yakir, M.; Srinivasan, S.; Nowak, J.; Izard, T.; Rangarajan, E.S.; et al. Structural and Molecular Mechanisms of Cytokine-Mediated Endocrine Resistance in Human Breast Cancer Cells. Mol. Cell. 2017, 65, 1122–1135.e5.
- Saw, C.L.; Wu, Q.; Kong, A.N. Anti-cancer and potential chemopreventive actions of ginseng by activating Nrf2 (NFE2L2) anti-oxidative stress/anti-inflammatory pathways. Chin. Med. 2010, 5, 37.
- Kwak, M.K.; Kensler, T.W. Targeting NRF2 signaling for cancer chemoprevention. Toxicol. Appl. Pharmacol. 2010, 244, 66–76.
- Kim, S.K.; Yang, J.W.; Kim, M.R.; Roh, S.H.; Kim, H.G.; Lee, K.Y.; Jeong, H.G.; Kang, K.W. Increased expression of Nrf2/ARE-dependent anti-oxidant proteins in tamoxifen-resistant breast cancer cells. Free Radic. Biol. Med. 2008, 45, 537–546.
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269.
- Kondo, N.; Toyama, T.; Sugiura, H.; Fujii, Y.; Yamashita, H. miR-206 expression is down-regulated in estrogen receptor α-positive human breast cancer. Cancer Res. 2008, 68, 5004–5008.
- Sachdeva, M.; Wu, H.; Ru, P.; Hwang, L.; Trieu, V.; Mo, Y.Y. MicroRNA-101-mediated Akt activation and estrogen-independent growth. Oncogene 2011, 30, 822–831.
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J. Biol. Chem. 2008, 283, 29897–29903.
- Payré, B.; de Medina, P.; Boubekeur, N.; Mhamdi, L.; Bertrand-Michel, J.; Tercé, F.; Fourquaux, I.; Goudounèche, D.; Record, M.; Poirot, M.; et al. Microsomal antiestrogen-binding site ligands induce growth control and differentiation of human breast cancer cells through the modulation of cholesterol metabolism. Mol. Cancer Ther. 2008, 7, 3707–3718.
- de Medina, P.; Silvente-Poirot, S.; Poirot, M. Tamoxifen and AEBS ligands induced apoptosis and autophagy in breast cancer cells through the stimulation of sterol accumulation. Autophagy 2009, 5, 1066–1067.
- Segala, G.; de Medina, P.; Iuliano, L.; Zerbinati, C.; Paillasse, M.R.; Noguer, E.; Dalenc, F.; Payré, B.; Jordan, V.C.; Record, M.; et al. 5,6-Epoxy-cholesterols contribute to the anticancer pharmacology of tamoxifen in breast cancer cells. Biochem. Pharmacol. 2013, 86, 175–189.
- Maggiore, R.J.; Gross, C.P.; Hurria, A. Polypharmacy in older adults with cancer. Oncologist 2010, 15, 507–522.
- Murphy, C.C.; Bartholomew, L.K.; Carpentier, M.Y.; Bluethmann, S.M.; Vernon, S.W. Adherence to adjuvant hormonal therapy among breast cancer survivors in clinical practice: A systematic review. Breast Cancer Res. Treat. 2012, 134, 459–478.
- Day, R.; National Surgical Adjuvant Breast and Bowel Projet P-1 study (NSABP-1). Quality of life and tamoxifen in a breast cancer prevention trial: A summary of findings from the NSABP P-1 study. National Surgical Adjuvant Breast and Bowel Project. Ann. N. Y. Acad. Sci. 2001, 949, 143–150.
- Polin, S.; Ascher, S. The effect of tamoxifen on the genital tract. Cancer Imaging 2008, 8, 135–145.
- Braithwaite, R.S.; Chlebowski, R.T.; Lau, J.; George, S.; Hess, R.; Col, N.F. Meta-analysis of vascular and neoplastic events associated with tamoxifen. J. Gen. Intern. Med. 2003, 18, 937–947.
- AlZaabi, A.; AlAmri, H.; ALAjmi, G.; Allawati, M.; Muhanna, F.; Alabri, R.; AlBusaidi, F.; AlGhafri, S.; Al-Mirza, A.A.; Al Baimani, K. Endometrial Surveillance in Tamoxifen and Letrozole Treated Breast Cancer Patients. Cureus 2021, 13, e20030.
- Palmer, J.L.; Trotter, T.; Joy, A.A.; Carlson, L.E. Cognitive effects of tamoxifen in pre-menopausal women with breast cancer compared to healthy controls. J. Cancer Surviv. 2008, 2, 275–282.
- Espeland, M.A.; Shumaker, S.A.; Limacher, M.; Rapp, S.R.; Bevers, T.B.; Barad, D.H.; Coker, L.H.; Gaussoin, S.A.; Stefanick, M.L.; Lane, D.S.; et al. Relative effects of tamoxifen raloxifene and conjugated equine estrogens on cognition. J. Womens Health 2010, 19, 371–379.
- Lee, K.C.; Ray, G.T.; Hunkeler, E.M.; Finley, P.R. Tamoxifen treatment and new-onset depression in breast cancer patients. Psychosomatics 2007, 48, 205–210.
- Blencowe, N.S.; Reichl, C.; Gahir, J.; Paterson, I. The use of Nolvadex in the treatment of generic Tamoxifen-associated small joint arthralgia. Breast 2010, 19, 243–245.
- Alekshun, T.; Patterson, S. Management of hot flashes in men with prostate cancer treated with androgen deprivation therapy. Support Cancer Ther. 2006, 4, 30–37.
- Perez, E.A. Safety profiles of tamoxifen and aromatase inhibitors in adjuvant therapy of hormone-responsive early breast cancer. Ann. Oncol. 2007, 18, 26–35.
- Avis, N.E. Breast cancer survivors and hot flashes: The search for nonhormonal treatments. J. Clin. Oncol. 2008, 26, 5008–5010.
- Cella, D.; Fallowfield, L. Recognition and management of treatment-related side effects for breast cancer patients receiving adjuvant endocrine therapy. Breast Cancer Res. Treat. 2008, 107, 167–180.
- Morrow, P.K.; Mattair, D.N.; Hortobagyi, G.N. Hot flushes: A review of pathophysiology and treatment modalities. Oncologist 2011, 16, 1658–1664.
- Cole, L.K.; Jacobs, R.L.; Vance, D.E. Tamoxifen induces triacylglycerol accumulation in the mouse liver by activation of fatty acid synthesis. Hepatology 2010, 52, 1258–1265.
- Jatobá, C.A.; Rezende, A.A.; Paiva Rodrigues, S.J.; Almeida Câmara, M.M.; das Graças Almeida, M.; Freire-Neto, F.; da Rocha, L.R.; da Medeiros, A.C.; Brandão-Neto, J.; Carvalho Formiga, M.C.; et al. Liver iron overload induced by tamoxifen in diabetic and non-diabetic female Wistar rats. Biometals 2008, 21, 171–178.
- Nguyen, M.C.; Stewart, R.B.; Banerji, M.A.; Gordon, D.H.; Kral, J.G. Relationships between tamoxifen use liver fat and body fat distribution in women with breast cancer. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 296–298.
- Nemoto, Y.; Saibara, T.; Ogawa, Y.; Zhang, T.; Xu, N.; Ono, M.; Akisawa, N.; Iwasaki, S.; Maeda, T.; Onishi, S. Tamoxifen-induced nonalcoholic steatohepatitis in breast cancer patients treated with adjuvant tamoxifen. Intern. Med. 2002, 41, 345–350.
- Ohnishi, T.; Ogawa, Y.; Saibara, T.; Nishioka, A.; Kariya, S.; Fukumoto, M.; Onishi, S.; Yoshida, S. CYP17 polymorphism and tamoxifen-induced hepatic steatosis. Hepatol. Res. 2005, 33, 178–180.
- Liu, C.L.; Yang, T.L. Sequential changes in serum triglyceride levels during adjuvant tamoxifen therapy in breast cancer patients and the Effect of dose reduction. Breast Cancer Res. Treat. 2003, 79, 11–16.
- Kim, Y.A.; Lee, S.; Jung, J.W.; Kwon, Y.J.; Lee, G.B.; Shin, D.G.; Park, S.S.; Yun, J.; Jang, Y.S.; Cho, D.H. Severe acute pancreatitis due to tamoxifen-induced hypertriglyceridemia with diabetes mellitus. Chin. J. Cancer Res. 2014, 26, 341–344.
- Baumgart, J.; Nilsson, K.; Evers, A.S.; Kallak, T.K.; Poromaa, I.S. Sexual dysfunction in women on adjuvant endocrine therapy after breast cancer. Menopause 2013, 20, 162–168.
- Tevaarwerk, A.J.; Wang, M.; Zhao, F.; Fetting, J.H.; Cella, D.; Wagner, L.I.; Martino, S.; Ingle, J.N.; Sparano, J.A.; Solin, L.J.; et al. Phase III comparison of tamoxifen versus tamoxifen plus ovarian function suppression in premenopausal women with node-negative, hormone-receptor positive breast cancer: A trial of Eastern Cooperative Oncology Group. J. Clin. Oncol. 2014, 32, 3948–3958.
- Carmassi, C.; Cordone, A.; Dell’Oste, V.; Pedrinelli, V.; Pardini, F.; Simoncini, M.; Dell’Osso, L. Prescribing Tamoxifen in Patients with Mood Disorders: A Systematic Review of Potential Antimanic Versus Depressive Effects. J. Clin. Psychopharmacol. 2021, 41, 450–460.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
794
Revisions:
2 times
(View History)
Update Date:
08 Jun 2023
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No